

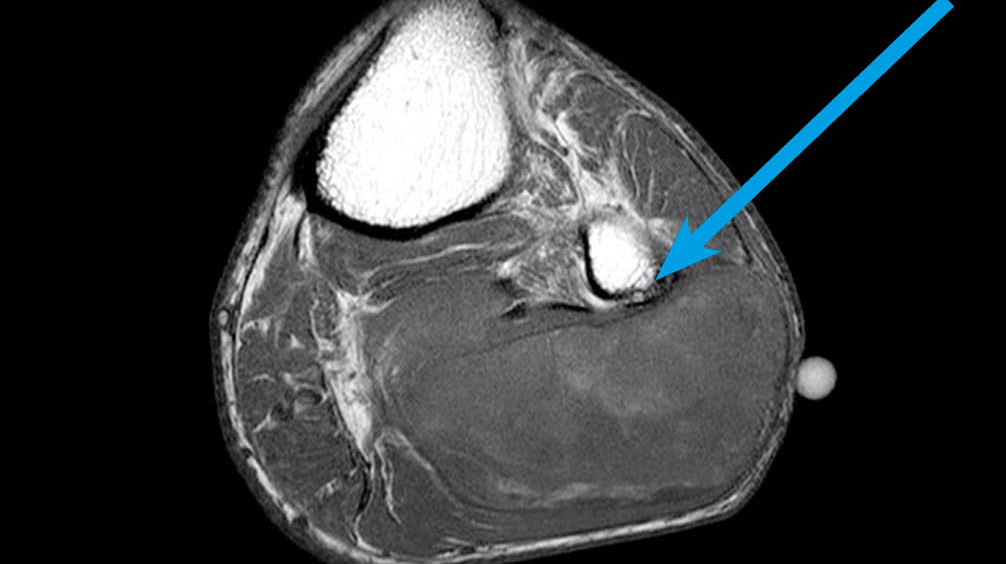
Der 85-jährige Patient habe seit mehr als 2 Jahren eine langsam zunehmende schmerzfreie Schwellung im proximal linken Unterschenkel bemerkt. Eine Vorstellung beim Hausarzt erfolgt erst als eine Fussheberparese aufgetreten ist. In der anschliessend durchgeführten MRT-Untersuchung des Unterschenkels wurde eine Raumforderung der Weichteile beschrieben. Eine weitere Abklärung hat der Patient initial abgelehnt. Wir werden im Artikel den Verlauf des spannenden Falles präsentieren sowie die Diagnostik und Therapie diskutieren.
Die initiale Vorstellung beim Hausarzt des 85-jährigen Mannes erfolgte aufgrund der Fussheberparese links. In der Anamnese erwähnte der Patient eine Schwellung des ipsilateralen Unterschenkels. Bei fehlenden Beschwerden hat der Patient diese Schwellung seinem Hausarzt bisher nicht rapportiert.

Klinisch zeigte sich eine glatte, rundliche, nicht druckdolente tumoröse Struktur im proximalen, posterioren Unterschenkel sowie eine Fussheberparese der ipsilateralen Seite. Die restliche klinische Untersuchung blieb bland. Angaben zur persönlichen Anamnese im Kasten. In der MRT-Untersuchung des linken Unterschenkels zeigte sich eine Raumforderung im postero-lateralen Muskelkompartiment in unmittelbarer Nachbarschaft zum N. peroneus (Abb. 1). Eine invasive Abklärung in Form von Biopsien hat der Patient abgelehnt. Zu den weiteren Nachkontrollen beim Hausarzt kam der Patient nicht mehr.
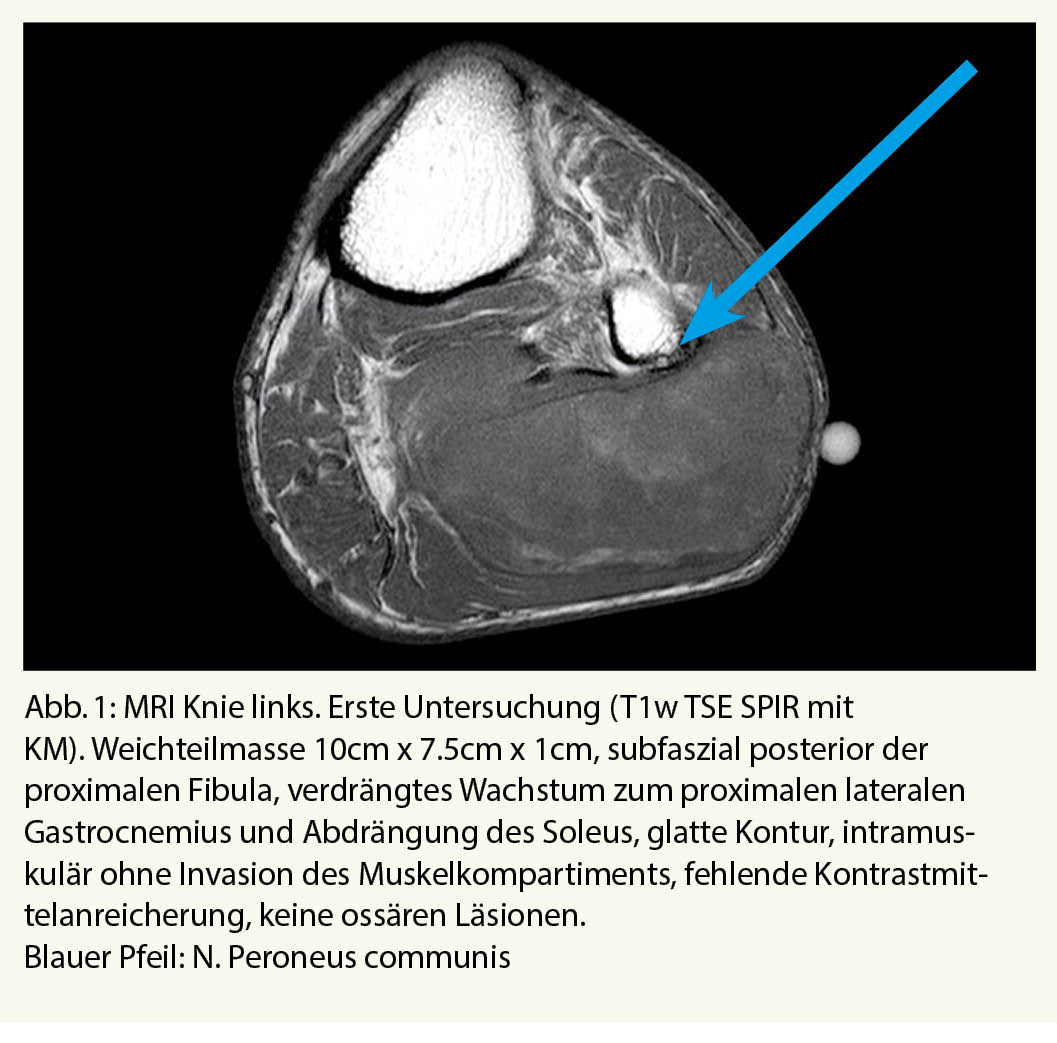
Mehr als 2 Jahre später erfolgte die erneute Vorstellung beim Hausarzt wegen der nun deutlich zunehmenden Schwellung am Unterschenkel. Jetzt konnte der Patient aufgrund der Schwellung nicht mehr die Hose anziehen. Gewichtsabnahme oder Nachtschweiss wurden verneint. Es wurde eine erneute MRT Untersuchung des Unterschenkels veranlasst (Abb. 2).
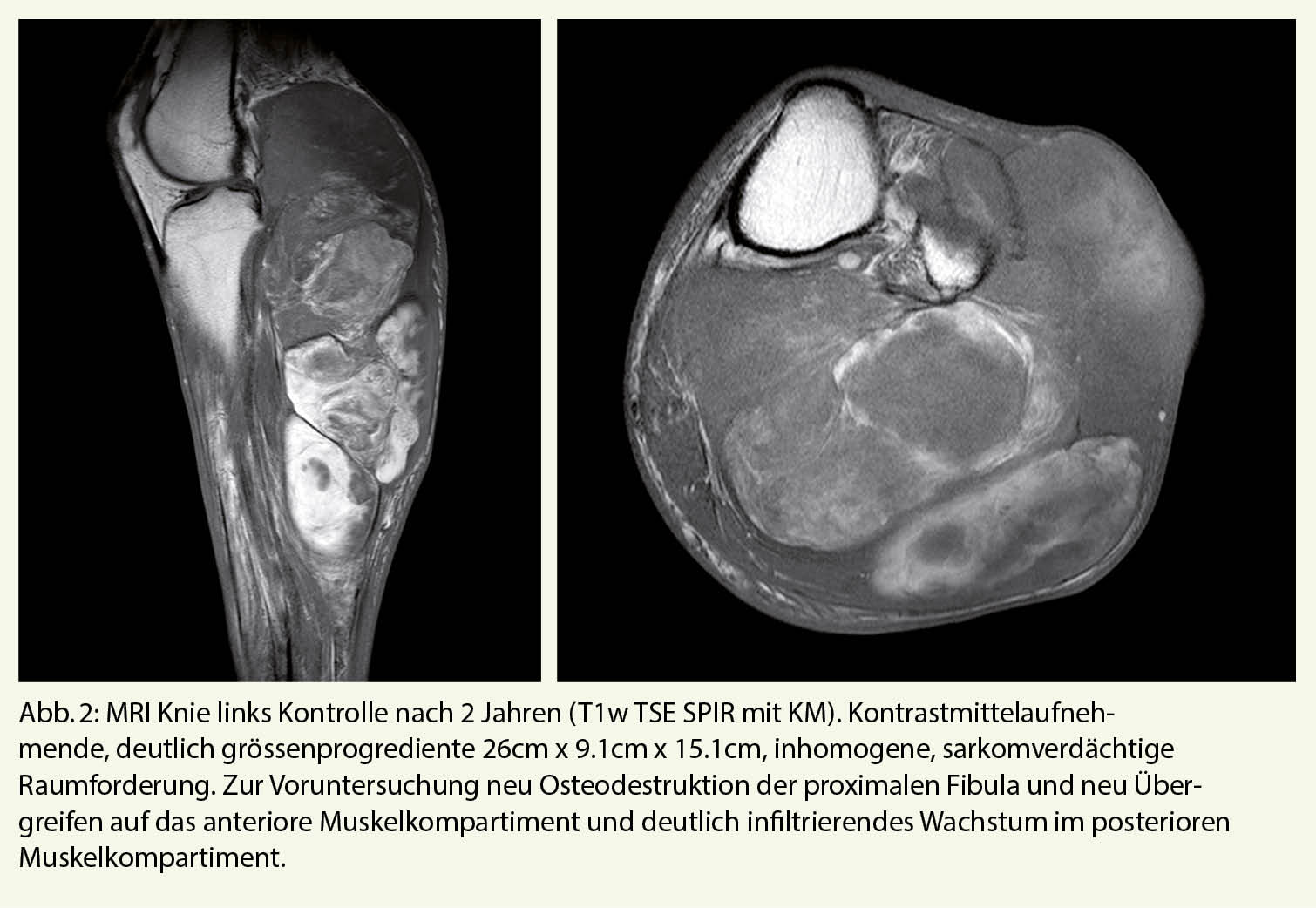
Status und Befunde
Bei der zweiten Vorstellung zeigte sich der Patient in gutem Allgemeinzustand. Lokal im linken Unterschenkel von der Poplitea bis zum distalen Unterschenkel zeigte sich eine grosse tumoröse Struktur mit intakter Haut und wenig Druckschmerz. Des Weiteren, seit 2 Jahren, vorbekannte paretisch bedinge Fussheberschwäche. Die regionalen Lymphknoten waren unauffällig. Das Labor blieb bland.
Diagnose und Therapie
Bei radiologisch hochverdächtigem Sarkom (inhomogene, grössenprogrediente und infiltrative Weichteilmasse) und nun bestehendem Einverständnis des Patienten für weiterführende Diagnostik erfolgte zunächst eine Stanzbiopsie. Das histopathologische Resultat zeigte ein Liposarkom von myxoidem Typ mit >5% rundzelliger Komponente.
Das Staging in Form eines CT Thorax und Abdomen blieb ohne Hinweis auf eine Metastasierung. Nach Besprechung des Falles im interdisziplinären Tumorboard wurde die Indikation für ein neoadjuvantes Therapiekonzept mit präoperativer lokaler Radiotherapie gefolgt von einer Tumorresektion in kurativer Intention gestellt.
Nach komplikationsloser Radiotherapie erfolgte eine vollständige Tumorresektion. Bei Nachweis lokaler Tumorinvasion erfolgte die zusätzliche Resektion von Fussbeuger, N. peroneus und Fibula um eine R0 Resektion zu erreichen. Die Fibularesektion erfolgte 5 cm ab dem fibulotibialen Gelenk, sodass das Ligamentum collaterale laterale belassen werden konnte. Die Peronaeus-Rekonstruktion erfolgte mit M. gastrocnemius-Plastik. In einer zweiten Sitzung erfolgte die Thierschplastik vom linken Oberschenkel auf den linken Unterschenkel.
Die histopathologische Untersuchung des Resektats ergab ein myxoides Liposarkom mit ausgedehnter rundzelliger Komponente (>35%), 10% Nekrosen und bis zu 28 Mitosen/10 HPF (FNCLCC-Grad 3), R0 Resektion, Tumordurchmesser ca. 260 mm. Daraus ergab sich folgende TNM Klassifikation: (Version 8 UICC [Union internationale contre le cancer]) pT4, pN0, M0, G3, R0, UICC Stadium IIIB.
Nach kurzer und komplikationsloser stationärer Rehabilitation konnte der Patient in gutem Allgemeinzustand entlassen werden. Eine Nachkontrolle mit CT Thorax und Abdomen wurde in 4 Monaten festgelegt. Der Patient konnte weiter in seinem häuslichen Umfeld selbstständig leben.
Verlauf
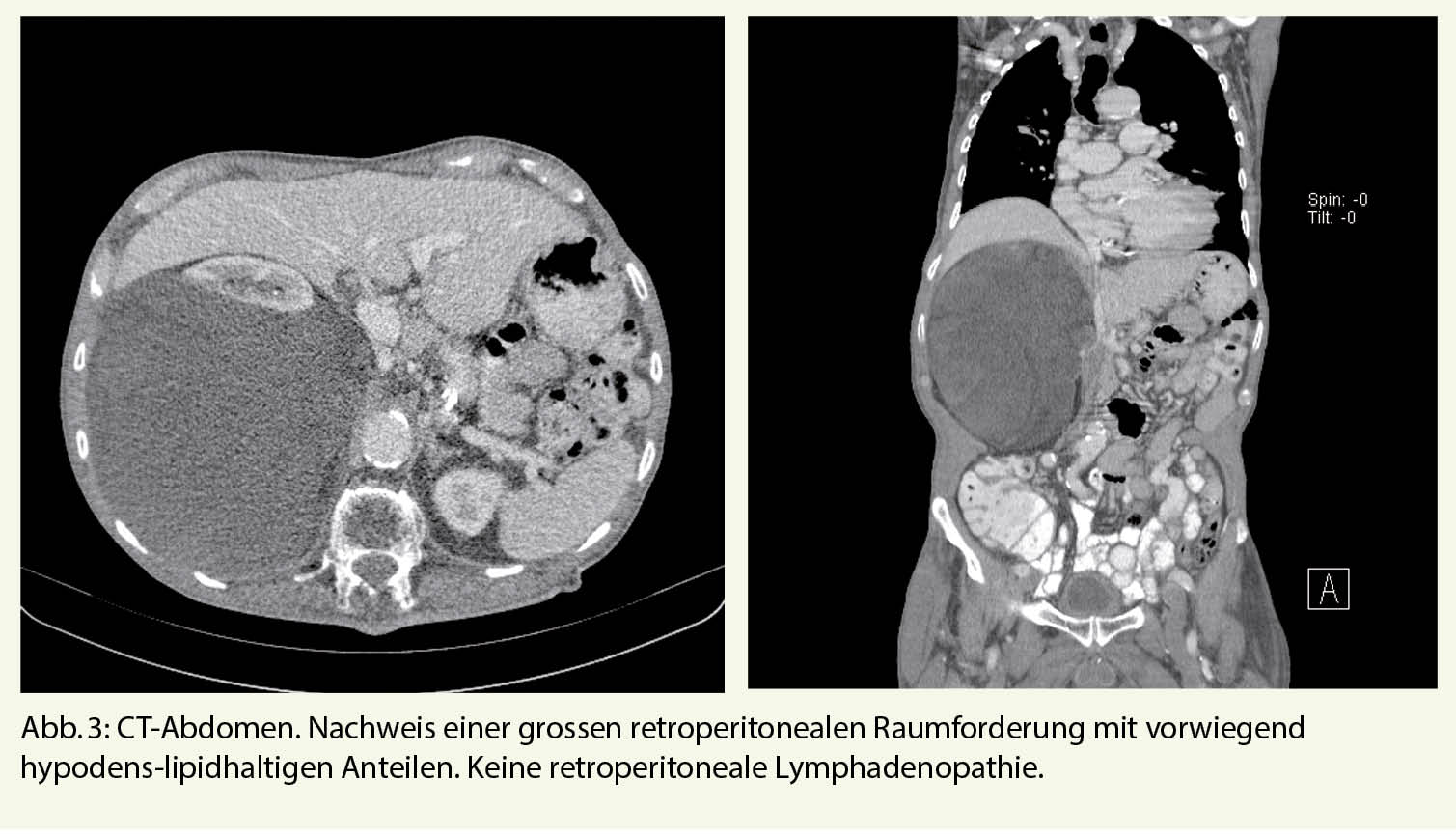
Drei Monate nach Austritt stellte sich der Patient bei seinem Hausarzt vor und beklagte rechtsseitige atemabhängige thorako-abdominale Schmerzen. Das CT Thorax/Abdomen wurde vorgezogen. Dies zeigte, neu zur Voruntersuchung, eine rechtsseitige grosse retroperitoneale Raumforderung mit vorwiegend lipomatösen Anteilen (Abb. 3). Weitere Abklärungen wie auch eine spezifische Therapie hat der Patient abgelehnt. Eine «best supportive care» Therapie mit peroraler Analgesie erfolgte mit Paracetamol und Metamizol bei Bedarf, worunter sich eine gute Schmerzkontrolle zeigte. Der Allgemeinzustand war noch gut.
Sieben Monate später wurde der Patient notfallmässig in das Spital eingewiesen. Der Patient war dehydriert und in kritischem Allgemeinzustand. Man beschränkte sich auf «best supportive care». Der Patient verstarb am achten Hospitalisationstag aufgrund von generalisiertem Infekt.
Diskussion
Weichteilsarkome haben eine Inzidenz von 4 - 5 pro 100 000 Einwohner pro Jahr in Europa (1). Das Liposarkom ist eines der häufigsten Weichteilsarkome. Seine Subtypen umfassen das pleomorphe, myxoide (-rundzellige), und dedifferenzierte Liposarkom. Davon abzugrenzen ist das gut-differenzierte Liposarkom oder aber atypischer lipomatöser Tumor. In fast allen Fällen entsteht das Sarkom aus normalen Adipozyten, welche sich vor allen in den Extremitäten sowie Retroperitoneum befinden, und nicht aus einer präexistenten Läsion (2).
Als Risikofaktoren gelten genetische Prädisposition, Radio- oder Chemotherapie, chemische Karzinogene, chronische Irritation und Lymphödem. Das Leitsymptom ist eine schmerzfreie wachsende tumoröse Struktur. Lokale typische Tumorinvasion ist selten. Metastasierung erfolgt vor allem hämatogen und meistens in die Lunge. Als Ausnahme metastasiert das Liposarkom vom myxoiden Typ typisch in das Retroperitoneum, Abdomen, Wirbelsäule und paravertebrale Weichteilgewebe und eher selten in die Lunge.
Diagnostik
Die Differentialdiagnose zwischen Lipom und Liposarkom ist schwierig. Lipome sind jedoch ca. 100x häufiger als Sarkome. Das Liposarkom zeigte sich in Frühstadien klinisch sowie radiologisch eher benigne und könnte als Lipom missdeutet werden. Weichteiltumoren müssen weiter abgeklärt werden, wenn eines der folgenden Kriterien erfüllt ist (3):
1. Subfasziale Tumorlokalisation
2. Weichteiltumor grösser als 5cm
3. Schwellung, welche an Grösse zunimmt
4. Schmerzhafte Schwellung
5. Rezidivschwellung nach Exzision
In einer prospektiven Studie mit 365 Patienten mit bestätigtem Weichteilsarkom, war die Tumorlokalisation (Kriterium 1) der sensitivste Indikator für Malignität. Es folgten die Grösse (>5 cm) und das Wachstum (4).
In unserem Fall hatte der Tumor bereits bei der ersten MRI Untersuchung eine Grösse > 5cm und eine Lage unmittelbar unterhalb der Muskelfaszien. Ausserdem konnte ein Wachstum dokumentiert werden. Aufgrund dieser Faktoren war eine Abklärung der Dignität mittels Core Nadel Biopsie (der heutige Standard) indiziert.
Die histopathologische Diagnostik soll gemäss der World Health Organisation (WHO) Klassifikation der Weichteiltumore von 2013 erfolgen. Der Malignitätsgrad soll in allen Fällen bestimmt werden. Hier wird das Grading System der «Federation Nationale des Centres de Lutte Contre le Cancer» (FNCLCC) verwendet.
Zum Staging wird das «Union internationale contre le cancer» (UICC) Klassifikationssystem verwendet. Das Staging muss ein Spiral-CT-Thorax enthalten. Bei myxoidem Liposarkom der Extremitäten muss zusätzlich ein Abdomen-CT durchgeführt werden. Szintigraphie, Ganzkörper MRI und PET Scan sind optional. Lymphknotenmetastasen sind selten.
Therapie
Die Standardtherapie des lokalisierten Sarkoms ist die Kombinationstherapie, bestehend aus präoperativer Strahlentherapie gefolgt von Chirurgie in R0 Resektion (5).
Über die Stellung der Chemotherapie besteht kein allgemeiner Konsens. Die Chemotherapie ist gegenwärtig nicht Teil einer Standardtherapie des lokalisierten Sarkomas, insbesondere nicht bei einem 85-jährigen Patienten.
Metastasen in den Lungen ohne extrapulmonalen Befall werden, falls möglich, chirurgisch behandelt. Extrapulmonale Metastasen, welche bei einem myxoiden Liposarkom nicht selten vorkommen, werden individuell behandelt. Bei Patienten mit myxoidem Liposarkom und solitärer extrapulmonaler Metastase bestehen die Möglichkeiten einer chirurgischen Entfernung sowie Ablation oder Radiotherapie.
Prognose
Prognostisch am wichtigsten sind histologischer Grad, Tumorgrösse und das pathologische Stadium zum Diagnosezeitpunkt. In einem Serien-Studium von «Memorial Sloan Kettering Cancer Center» (MSKCC) betrug die 5-Jahre Überlebensrate zwischen 86% (TNM Stadium I) und 52% (TNM Stadium III) (6).
High risk Patienten haben ein Rezidiv in den ersten 2 - 3 Jahren. Low risk Patienten entwickeln, wenn überhaupt, erst spät ein Rezidiv. Dem endsprechend sollten high risk Patienten in den ersten 2 - 3 Jahren alle 3 - 4 Monate mittels CT/MRI nachkontrolliert werden. Danach sollten weitere Kontrollen im 6-monatigen Intervall erfolgen. Ab dem 5. Jahr sind jährliche Kontrollen indiziert. Low Risk Patienten sollten alle 4 - 6 Monate mittels konventionellem Thoraxröntgen oder Thorax-CT in den ersten 3 - 5 Jahren nachkontrolliert werden und dann weiter jährlich. Im Gegensatz zu den anderen Sarkomen, sollen die Nachkontrollen beim myxoiden Liposarkom zusätzlich ein Abdomen-CT enthalten wegen der atypischen Metastasierung in das Retroperitoneum/Abdomen.
Copyright bei Aerzteverlag medinfo AG
Peter Rot-Strasse 113
4058 Basel
Dr.Lantcron@gmail.com
Kantonsspital Baselland Laufen
Lochbruggstrasse 39
4242 Laufen
Die Autoren haben im Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019; 69:7.
2. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. World Health Organization Classification of tumours of soft tissue and bone, 4th ed, IARC Press, Lyon 2013
3. Sinha S, Peach AH. Diagnosis and management of soft tissue sarcoma. BMJ 2010 Dec 29;341
4. Hussein R, Smith MA. Soft tissue sarcomas: are current referral guidelines sufficient? Ann R Coll Surg Engl 2005; 87:171.
5. ESMO/European Sarcoma Network Working Group. Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014; 25 Suppl 3:iii102.
6. American Joint Committee on Cancer Staging Manual, Edge SB, Byrd DR, Compton CC, et al (Eds), Springer, New York 2010. p.291.


der informierte @rzt
- Vol. 10
- Ausgabe 10
- Oktober 2020