Die pulmonale Hypertonie (PH) ist eine chronisch verlaufende Erkrankung, welche alleine oder in Assoziation mit anderen Krankheiten vorkommen kann und unbehandelt eine hohe Mortalität aufweist. Das Leitsymptom der PH ist die anstrengungsabhängige Atemnot. Eine Vielzahl der Patienten wird aufgrund der anfänglich in Ruhe fehlenden und unspezifischen Symptomen erst spät diagnostiziert, die Beschwerden werden von Patienten und behandelnden Ärzten häufig zuerst auf «Trainingsmangel» zurückgeführt.
Die aktuellen Empfehlungen zur Klassifikation, Diagnostik und Therapie basieren auf den gemeinsamen Guidelines der ERS und ESC, die im Jahr 2015 veröffentlicht wurden und den Proceedings aus der PH-Weltkonferenz in Nizza 2018 (1-3).
Klassifikation
Die pulmonale Hypertonie wird nach den immer noch geltenden ERS/ESC-Guidelines definiert als ein gesteigerter mittlerer pulmonal-arterieller Druck (mPAP) ≥ 25 mmHg in Ruhe, invasiv ermittelt in einer Rechtsherzkatheter-Untersuchung. Normalerweise beträgt der mPAP in Ruhe 14 + /- 3 mmHg mit einer geschätzten oberen Grenze von ca. 20 mmHg (4). Deshalb wurde in der internationalen Expertenkonferenz in NIZZA 2018 vorgeschlagen, die Schwelle auf > 20 mmHg zu senken (2).
Zur weiteren hämodynamischen Unterteilung erfolgt die invasive Messung des «gemittelten» pulmonal arteriellen Wedge-Druckes (PAWP) (Tab. 1). So ist eine «präkapilläre PH» definiert als PAWP ≤ 15 mmHg. Nach dem Weltsymposium in Nizza beinhaltet die neu vorgeschlagene Definition der präkapillären PH neben dem mPAP > 20mmHg und dem PAWP ≤ 15 mmHg neu auch einen pulmonal-vaskulären Widerstand (PVR) ≥ 3 WU.
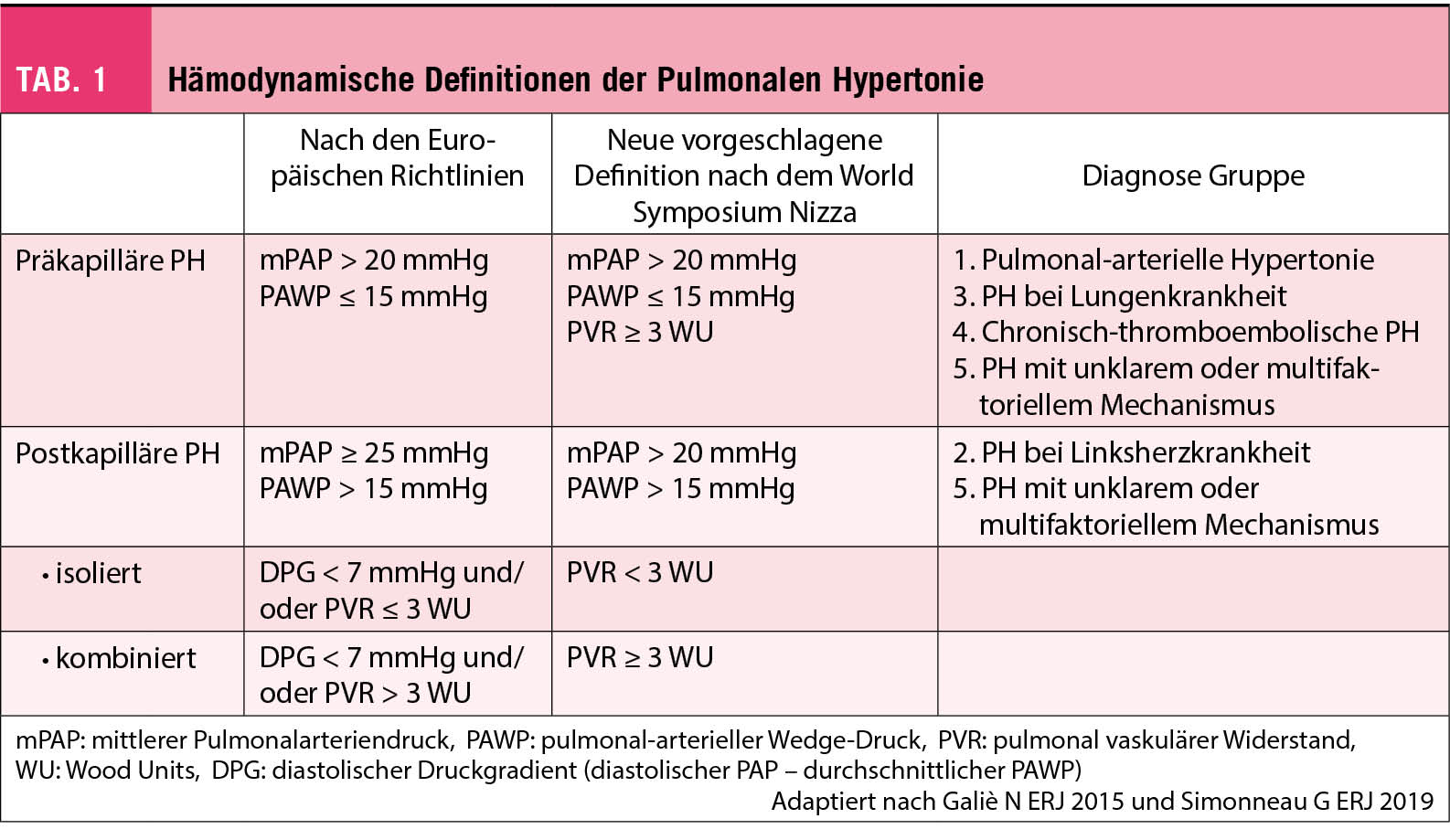
In die Gruppe der präkapillären PH fallen die pulmonal-arterielle Hypertonie (PAH), PH aufgrund von Lungenerkrankungen, die chronisch-thromboembolische PH sowie die PH mit unklarem oder multifaktoriellem Mechanismus (Tab. 2 und 3).
Bei Vorliegen eines PAWP > 15 mmHg spricht man von einer «postkapillären PH». Diese kommt im Rahmen von Linksherzerkrankungen vor. Auch hier wurde neu vorgeschlagen, die Grenze auf > 20mmHg zu senken (2). Die postkapilläre PH wird anhand des PVR weiter unterteilt in die «isolierte postkapilläre» bei einen PVR < 3 WU und in die «kombinierte postkapilläre und präkapilläre PH» bei einem PVR ≥ 3 WU.
Die klinische Unterteilung erfolgt in fünf Gruppen («Nizza-Klassifikation»), die in den Tabellen 2 und 3 aufgelistet sind.
Inwieweit somit ein mPAP zwischen 21 und 24 mmHg oder ein PVR zwischen 2-3 WU klinisch relevant ist, ist noch nicht gänzlich geklärt. Dieser Grenzbereich wird auch «Borderline-Erhöhung» genannt und spielt vor allem für Patienten eine Rolle, die einem erhöhten Risiko unterliegen eine manifeste PAH zu entwickeln. Dazu zählen v.a. Patienten mit einer Kollagenose, insbesondere der Sklerodermie, oder Familienangehörige von Patienten mit hereditärer PAH. In diesen Risikogruppen sollten Verlaufskontrollen konsequent erfolgen. Eine frühe medikamentöse Therapieeinleitung wird generell empfohlen (5).
Zur Definition einer Belastungs-induzierten pulmonalen Hypertonie fehlen verlässliche Daten, insbesondere zur Klassifikation und Prognose, weshalb diese Entität nicht in den PH-Leitlinien aufgenommen wurde. Es konnte aber bereits nachgewiesen werden, dass es unter Belastung zu einem abnormen pulmonalen Druckanstieg im Verhältnis zum Fluss kommt. Dieser starke pulmonale Druckanstieg kann auf eine Erhöhung des PVR sowie des links-atrialen Drucks zurückzuführen sein. Eine «belastungsinduzierte pulmonale Hypertonie» könnte laut Studien bei einem mPAP ≥ 30 mmHg, einem Herzminutenvolumen < 10 l/min resp. mPAP/CO > 3 mmHg/l/min vorliegen/definiert werden (6).


Diagnostik
Die klinische Präsentation ist abhängig vom Schweregrad der Erkrankung. Das Leitsymptom ist die Belastungsdyspnoe, für welche bis anhin keine Ursache gefunden werden konnte und die reduzierte körperliche Leistungsfähigkeit. In fortgeschrittenen Stadien kann es zu Synkopen, Thoraxdruck (ähnlich der Angina pectoris), Rhythmusstörungen und Beinödemen bis hin zum Rechtsherzversagen kommen. Ein Leitbefund ist die Sauerstoffdesaturation unter Belastung, elektro- und echokardiografische Zeichen der Rechtsherzbelastung und ein erhöhtes (NT-pro-)BNP. Daneben können auch der Nachweis einer tiefen Diffusionsstörung in der Lungenfunktion sowie typische Befunde in der Spiroergometrie hinweisend für eine pulmonale Drucksteigerung sein und eine weitere Differenzierung der Ätiologie erlauben (7). In der Diagnostik soll auch ein klinisches und laborchemisches Autoimmun-Screening zum Nachweis einer rheumatologischen Grunderkrankung, ein HIV-Test, eine Leberwert- und Sonografie-Untersuchung, die Ventilations-Perfusions-Szintigraphie zur Suche einer möglichen CTEPH sowie das HRCT-Thorax zur Suche einer zugrundeliegenden Lungenparenchymerkrankung (z.B. ILD, Emphysem) durchgeführt werden (7).
Durch die Beurteilung des funktionellen Status, der Leistungsfähigkeit, des hämodynamischen Schweregrades und den Resultaten weiterer apparativer Diagnostik mittels Echokardiographie, Spiroergometrie, NT-pro-BNP kann eine Risikostratifizierung erfolgen (Tab. 4). Da das Mortalitätsrisiko sehr hoch ist, sollte diese in regelmässigen Abständen erfolgen. Daraus ergeben sich wichtige Informationen zur Prognose resp. Mortalität mit daraus resultierenden diagnostischen Massnahmen und Therapieanpassungen bei Krankheitsprogression. Ziel ist das Erreichen eines niedrigen Mortalitätsrisikos mit einer Verbesserung der körperlichen Leistungsfähigkeit, Lebensqualität und RV-Funktion.
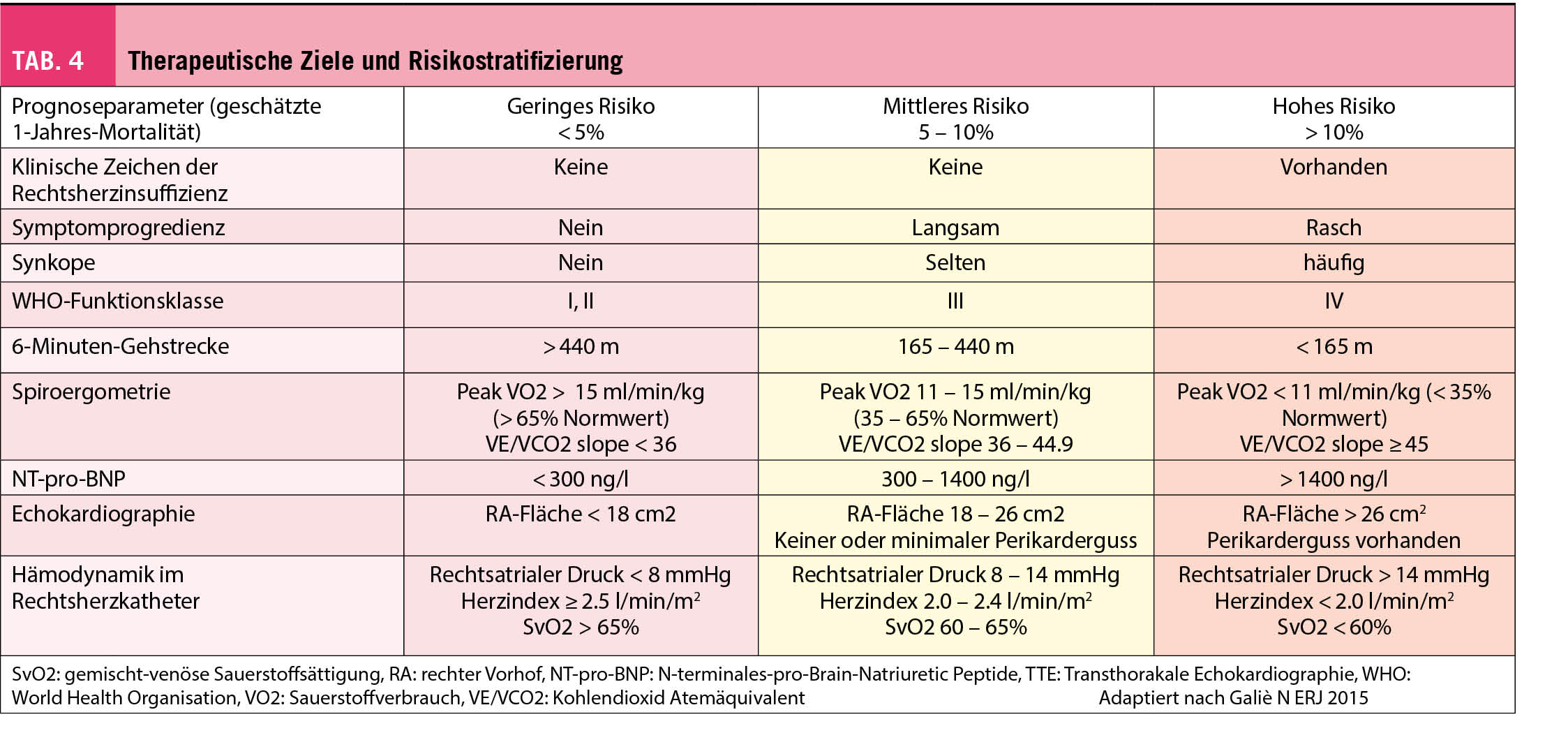
Die Echokardiographie gilt als wichtigste nicht invasive Screeningmethode zur Detektion einer pulmonalen Drucksteigerung (7).
Gemäss ESC/ERS-Leitlinien wird empfohlen, die Wahrscheinlichkeit des Vorliegens einer PH anhand der Trikuspidalklappen-Regurgitationsgeschwindigkeit und anderer echokardiographischer Hinweise in hoch, intermediär und gering zu graduieren, um eine Rechtsherzkatheter- Untersuchung zur Bestätigung zu veranlassen.
Hierbei ist aber zu bedenken, dass die Ergebnisse der Echokardiographie Untersucher- und Patientenkonstitutionsabhängig variieren und in 10-30 % der Fälle die Diagnose trotz symptomatischen Patienten entgeht (1, 8). Deshalb sollte bei ungeklärter Dyspnoe nichtsdestotrotz die Verdachtsdiagnose einer PH in Betracht gezogen werden. Die Rechtherzkatheter-Untersuchung gilt in jedem Fall als «Goldstandard» zur Diagnose einer PH und sollte, aufgrund des niedrigen Risikos insbesondere bei Risikopatienten niederschwellig durchgeführt werden. Zu beachten ist hier jedoch, dass diese Untersuchung sehr komplex ist und daher in einem erfahrenen Zentrum durchgeführt werden soll. Idealerweise erfolgen Messungen der hämodynamischen Parameter inklusive repetitiver Bestimmung des Herzauswurfes mittels direktem Fick oder Thermodilution in Ruhe und bei Belastung. Bei jedem Patienten mit V.a. PH sollte dringend eine Vasoreagibilitätsmessung (mittels NO / Stickstoffmonoxid oder Prostazyklinen) durchgeführt werden, da dies für die Therapie eine klare Konsequenz hat. Responder (Abfall des mPAP < 40 mmHg, und um 10 mmHg im Vergleich zum Ausgangswert, bei unveränderten oder angestiegenen Herzzeitvolumen) müssen mit einem Kalziumantagonisten behandelt werden (9). Es ist jedoch sehr wichtig, dass diese Patienten regelmässig im PH-Zentrum nachkontrolliert werden, um die dauerhafte Response sicherzustellen.
PH-spezifische Therapie
Medikamentös
Die gezielte Therapie bei Vorliegen einer PAH erfolgt bei Nachweis einer Vasoreagibilität mit Kalziumantagonisten; bei Fehlen einer Vasoreagibilität mit Phosphodiesterase-5-Hemmer (Sildenafil, Tadalafil), Stimulatoren der löslichen Guanylatcyclase (Riociguat), Endothelin-Rezeptor-Antagonisten (Ambrisentan, Bosentan, Macitentan), parenteralen Prostazyklinen (Epoprostenol, Iloprost, Treprostinil) oder einem oralen Prostazyklin-Rezeptor-Agonist (Selexipag). Kombinationstherapien werden bereits von Beginn an aufgrund nachgewiesener Verbesserung des funktionellen Status und verzögerter Verschlechterung und dadurch wahrscheinlich verbessertem Überleben empfohlen (9).
Nebenwirkungen dieser Medikamente beinhalten Kopfschmerzen, gastrointestinale Beschwerden, Arthralgien, Myalgien, Kieferschmerzen und Veränderungen der Blutwerte, weshalb diese komplexen Kombinationstherapien nur in enger Zusammenarbeit mit einem PH-Zentrum erfolgen sollten.
Die Therapie der Wahl bei Vorliegen einer CTEPH ist, falls die Lungengefässveränderungen technisch operabel sind, die pulmonale Endarteriektomie (10). Bei Inoperabilität werden Patienten medikamentös und/oder mit einer interventionellen Ballon-Angioplastie behandelt. Die Ultima ratio bei therapierefraktärem Verlauf einer PAH bleibt die Lungen-Transplantation.
Die Wertigkeit einer Therapie mit PH-spezifischen Medikamenten bei «Borderline-PH», einer nur belastungsinduzierten PH ist aktuell mangels Evidenz ausserhalb von Forschungsprojekten nicht empfohlen, dasselbe gilt für die hochprävalenten Gruppen der PH bei Linksherz- oder Lungenkrankheiten. Umso wichtiger ist es, die Datenlage diesbezüglich zu verbessern und Patienten, wenn immer möglich, an ein spezialisiertes Zentrum mit der Möglichkeit von Studien zuzuweisen.
Allgemein
Regelmässige körperliche Aktivität ist für die Lebensqualität und für den allgemeinen Krankheitsverlauf sehr positiv, sollte aber unbedingt dosiert Symptom-orientiert und gegebenenfalls auch im Rahmen einer stationären Rehabilitation bei Dekonditionierung erfolgen. In der Schweiz wurde ein spezifisches PH-Rehabilitationsprogramm in der Klinik Barmelweid unter Anleitung des Europäischen Pionier-Rehabilitationsprogramms in Heidelberg implementiert. Exzessives Training sollte vermieden werden (11). Schwangerschaften bei PAH sind mit einer deutlich erhöhten Mortalitätsrate assoziiert und sind, wenn überhaupt, nur bei unter Therapie im Alltag normal leistungsfähigen Patientinnen mit normalisierter Hämodynamik unter engmaschiger interdisziplinärer Begleitung im zuständigen PH-Zentrum möglich. Dem Grossteil der Patientinnen muss jedoch dringend abgeraten werden (11). Aufgrund des erhöhten Operationsrisikos sollten Eingriffe, wann immer möglich, in lokaler oder epiduraler Anästhesie in einem Zentrumsspital mit Erfahrung und der Möglichkeit einer ECMO, Herzanästhesie und spezifischen intensivmedizinischen Betreuung erfolgen (11).
Die Empfehlungen einer oralen Antikoagulation bei PAH sind nicht eindeutig und teilweise kontrovers, bei PAH assoziiert mit Sklerodermie klar nicht empfohlen. Bei IPAH, hereditärer und medikamentös-toxischer PH wird im Einzelfall nach Nutzen- Risiko-Abwägung entschieden, ob diese erfolgen soll.
Die Indikation zur oralen Antikoagulation beim Vorliegen einer CTEPH ist lebenslang gegeben (Ziel INR 2-3), selbst nach erfolgreicher chirurgischer Therapie. Dabei kommen Kumarin-Derivate oder andere orale Antikoagulation in Frage, wobei für die Wahl welcher Antikoagulation die Datenlage zur Behandlung der CTEPH gering ist (11).
Bei diesem Artikel handelt es sich um einen Zweitabdruck des in «der informierte arzt» 08-2020 erschienen Originalartikels.
Copyright bei Aerzteverlag medinfo AG
Klinik für Pneumologie
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
diana.mandler@usz.ch
Die Autoren haben in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
1. Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 2015;46:903-75.
2. Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019;53.
3. Rosenkranz S, Diller GP, Dumitrescu D, et al. [Hemodynamic Definition of Pulmonary Hypertension: Commentary on the Proposed Change by the 6th World Symposium on Pulmonary Hypertension]. Dtsch Med Wochenschr 2019;144:1367-72.
4. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J 2009;34:888-94.
5. Galie N, Channick RN, Frantz RP, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2018.
6. Kovacs G, Herve P, Barbera JA, et al. An official European Respiratory Society statement: pulmonary haemodynamics during exercise. Eur Respir J 2017;50.
7. Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J 2019;53.
8. Coghlan JG, Denton CP, Grunig E, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014;73:1340-9.
9. Hoeper MM, Apitz C, Grunig E, et al. Targeted therapy of pulmonary arterial hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int J Cardiol 2018;272S:37-45.
10. Wilkens H, Konstantinides S, Lang IM, et al. Chronic thromboembolic pulmonary hypertension (CTEPH): Updated Recommendations from the Cologne Consensus Conference 2018. Int J Cardiol 2018;272S:69-78.
11. Grunig E, Benjamin N, Kruger U, et al. General measures and supportive therapy for pulmonary arterial hypertension: Updated recommendations from the Cologne Consensus Conference 2018. Int J Cardiol 2018;272S:30-6.

info@herz+gefäss
- Vol. 10
- Ausgabe 5
- Oktober 2020