Aufgrund der raschen Entwicklung im molekular-genetischen Verständnis primärer Hirntumoren wird 2021 eine Revision der WHO Klassifikation publiziert. Dort finden sich wesentliche Neuerungen zum diagnostischen Algorithmus und zur Nomenklatur primärer Hirntumoren. Wir haben in der Ausgabe 03-21 der «info@onkologie« darüber berichtet. Therapeutische Anpassungen an die neue Klassifikation sind Thema des folgenden Beitrags.
IDH1– und viel seltener IDH2-Mutationen sind frühe Ereignisse in der Gliomentwicklung. Biologisch und prognostisch unterscheiden sich Gliome dahingehend, ob sie mit oder ohne diese Mutationen auftreten und auch die Therapiestruktur richtet sich danach. Wir gliedern deshalb im Folgenden die Gliome in die beiden Gruppen «IDH-mutiert» und «IDH-wild-type».
Therapie IDH-mutierter diffuser Gliome
Hintergrund:
Mutationen im Gen für Isozitrat-Dehydrogenase (IDH) sind mit bis zu 80 % die weitaus häufigste genetische Veränderung diffuser Gliome der WHO-Grade 2-4. Sie unterscheiden sich in Bezug auf den natürlichen Verlauf und das Therapieansprechen von IDH-wild-type Gliomen. In klinischen Studien werden Therapien gegen mutiertes IDH in Form von spezifischen Inhibitoren oder Impfungen mit dem Peptid, welches die charakteristische Mutation enthält, geprüft (Abb. 1).
IDH-Mutationen treten in Gliomen überwiegend in Form einer IDH1R132H-Mutation auf. Sie führen zu einer neomorphen, d.h. Tumor definierenden enzymatischen Funktion des IDH-assoziierten Proteins mit Konversion von α-Ketoglutarat (α-KG) zu 2-Hydroxyglutarat (2-HG).
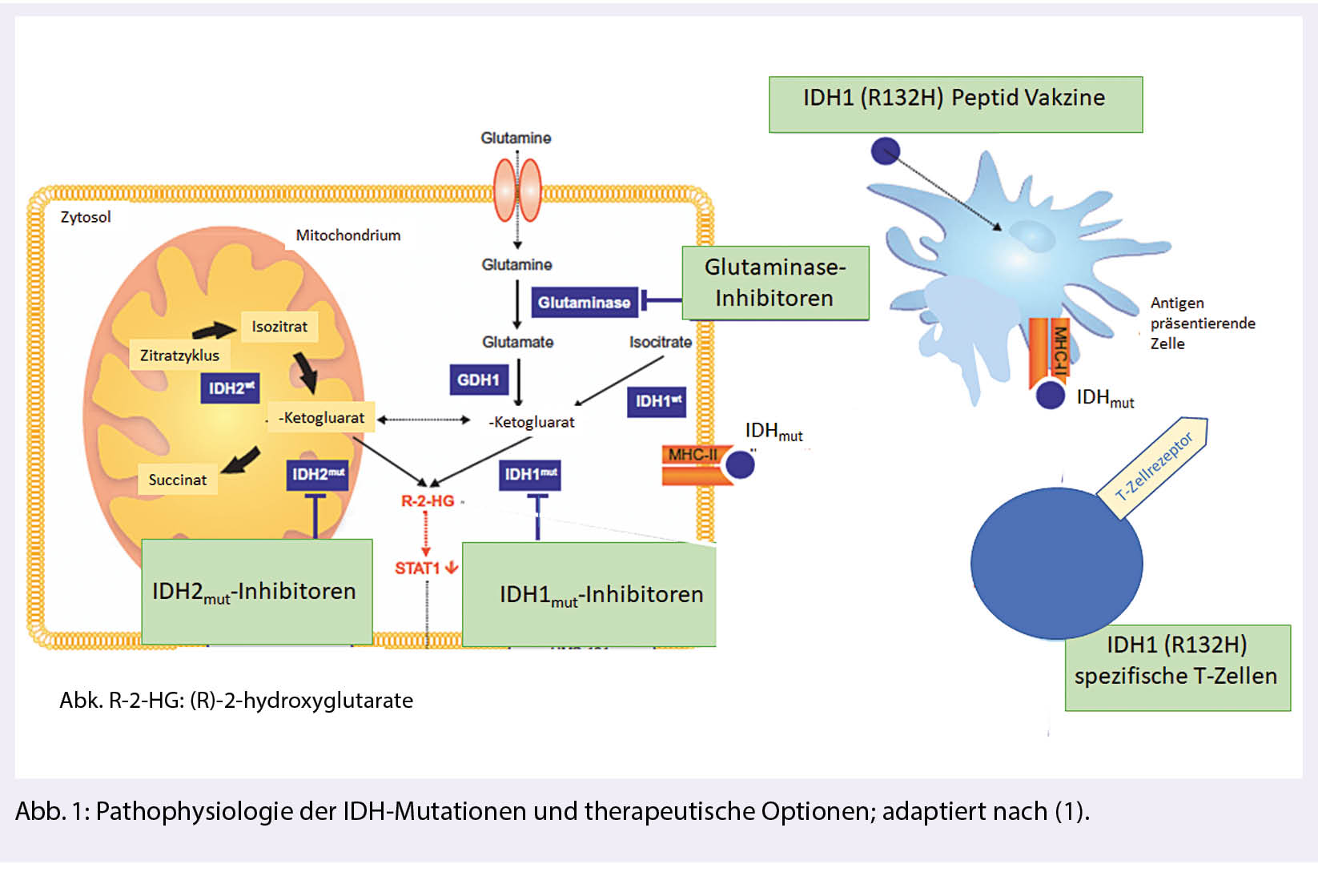
Der Onkometabolit 2-HG akkumuliert in Tumorzellen und führt über verschiedene epigenetische Mechanismen zu genomischer Instabilität, Akkumulation weiterer Mutationen und maligner Transformation.
IDH-Mutationen werden in der klinischen Routine anhand eines mutationsspezifischen Antikörpers, welcher die häufige IDH1R132H-Mutation erkennt, immunhistochemisch diagnostiziert. Bei fehlender Reaktivität und Vorliegen eines diffusen Glioms ist eine Sequenzierung erforderlich, um das Vorliegen seltener IDH1– oder IDH2-Mutationen auszuschliessen. Viele Institutionen machen dies bis zu einem Patientenalter von 60 Jahren, da darüber hinaus kaum IDH-mutierte Gliome zu erwarten sind. Indirekte Hinweise für das Vorliegen einer IDH-Mutation können Methylierungs-Arrays liefern, die eine charakteristische, durch die Akkumulation von 2-HG bedingte globale DNA-Hypermethylierung anzeigen. Mit der 2-HG-Magnetresonanzspektroskopie ist ein Verfahren in Entwicklung, das eine nicht-invasive bildgebende Diagnose von IDH-mutierten Gliomen ermöglichen soll.
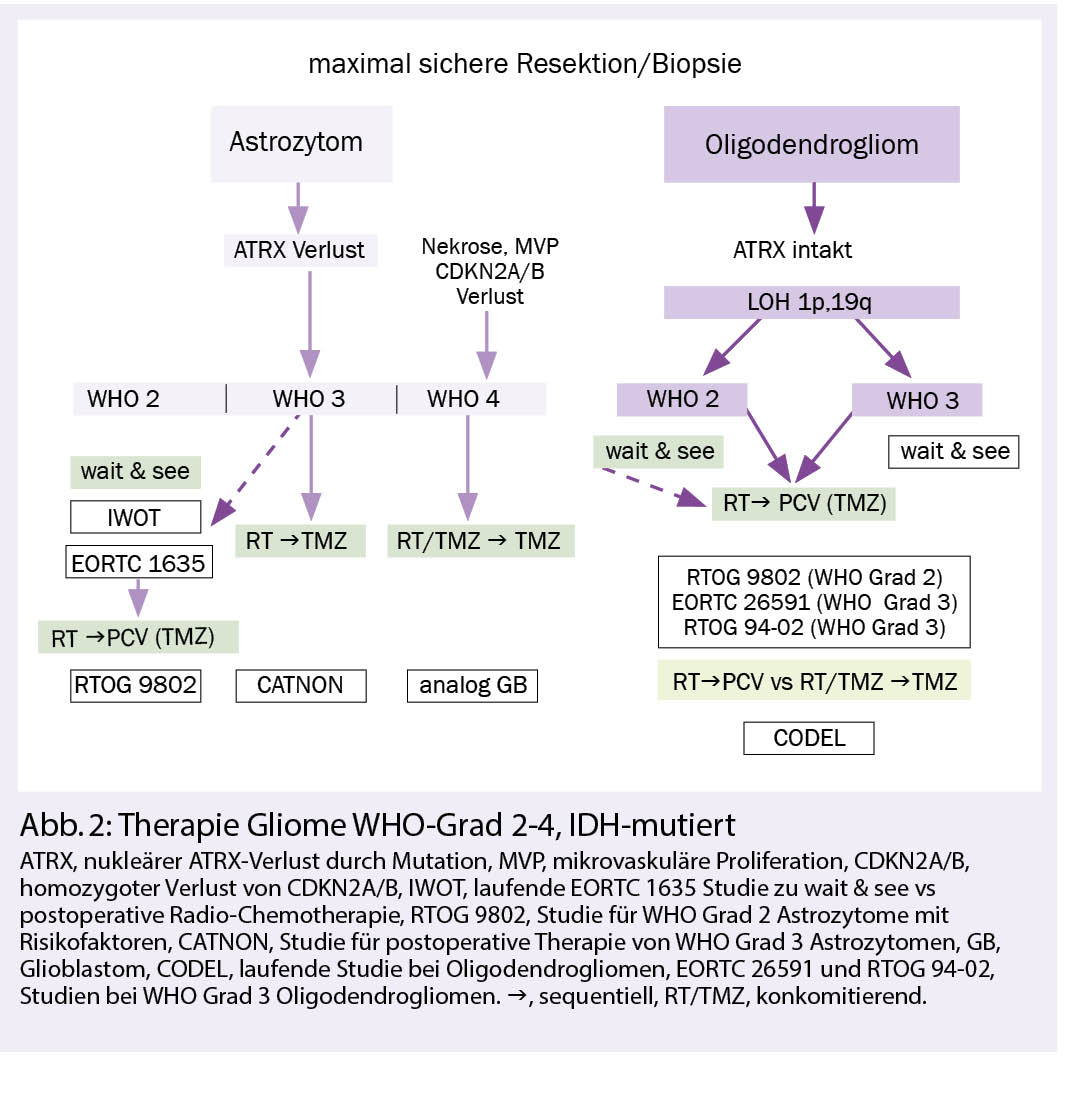
In der grossen Gruppe IDH-mutierter Gliome wird eine zunehmende Zahl molekular definierter Subtypen mit Bedeutung für Prognose und Therapiewahl zusammengefasst. IDH-mutierte Astrozytome weisen einen nukleären Verlust des Transkriptionsregulators ATRX auf und unterscheiden sich damit von den IDH-mutierten Oligodendrogliomen, die ihrerseits einen kombinierten Allelverlust auf den Chromosomen 1p und 19q aufweisen. Innerhalb der IDH-mutierten Astrozytome ist das Vorliegen von mikrovaskulären Proliferaten, Nekrosen und insbesondere der homozygote Verlust von CDKN2A/B mit einer ungünstigen Prognose assoziiert und wird dem WHO-Grad 4 zugeordnet. Diese Tumoren werden somit nicht mehr als Glioblastome bezeichnet, sondern als «Astroyztom, IDH-mutiert, WHO-Grad 4».
Therapie WHO Grad 2 und 3, IDH-mutierte Gliome:
Nach bildgebender Verdachtsdiagnose erfolgt eine möglichst vollständige und neurologisch schonende Resektion und eine integrierte neuropathologische Diagnose. Diese beinhaltet die histomorphologische und molekulargenetische Gewebeanalyse, welche auch anhand einer stereotaktischen Biopsie gestellt werden kann, wenn eine umfangreichere Resektion nicht indiziert ist. Zunehmend wird bei den Oligodendrogliomen die klinische Bedeutung des WHO-Grades 2 versus 3 in Frage gestellt. Dabei ist allerdings nicht geklärt, was dies für die zu wählende Radiotherapiedosis bedeutet, da es hierfür keine Studiendaten gibt.
Astrozytome und Oligodendrogliome WHO-Grad 2 können bei Fehlen der zwei wichtigsten Prognose bestimmenden Risikofaktoren (RF) – nämlich unvollständige Resektion und Alter über 40 Jahre – zunächst beobachtet werden. Basierend auf den Daten grosser randomisierter Studien wird bei Progress oder Vorhandensein der genannten RF eine Radiotherapie mit 50.4-54 Gy, gefolgt von Procarbazin, CCNU und Vincristin (PCV-Schema) für 6 Zyklen empfohlen oder alternativ, jedoch nicht evidenzbasiert, mit Temozolomid (2, 3).
Die Strahlentherapie bleibt nach wie vor Teil der Standardtherapie bei WHO-Grad-2-Gliomen. In einer randomisierten Phase-III-Studie (EORTC 22033-26033) war die Radiotherapie im Vergleich zur alleinigen Chemotherapie in der molekularen Subgruppenanalyse bei IDH-mutierten Astrozytomen ohne 1p,19q Ko-Deletion der alleinigen Chemotherapie mit Temozolomid über ein Jahr hinsichtlich des progressionsfreien Überlebens überlegen (4).
Die Frage nach dem optimalen Zeitpunkt einer Radio-Chemotherapie bei Grad 2-(und 3-) Astrozytomen ohne LOH 1p,19q ist nicht abschliessend geklärt. Das Abwägen einer postoperativen Tumorkontrolle gegenüber dem Auftreten von Toxizität nach Radiotherapie soll in der laufenden EORTC 1635 IWOT-Studie geklärt werden (NCT03763422). Die Studie vergleicht die postoperative Radio-Chemotherapie mit einer aktiven Nachsorge.
Bei Oligodendrogliomen WHO-Grad 3 belegen Langzeitdaten aus zwei randomisierten Phase-III-Studien (RTOG 9402 und EORTC 26951) den Benefit einer postoperativen Radiotherapie mit 59.4 Gy üblicherweise gefolgt von 6 Zyklen PCV oder wie in der RTOG-Studie mit 4 Zyklen PVC vor der Radiotherapie (5, 6.). Die noch nicht abgeschlossene, internationale CODEL-Studie (NCT00887146) vergleicht bei Oligodendrogliomen in einem Nichtunterlegenheits-Design eine sequenzielle Radiochemotherapie mit PCV (bis 6 Zyklen) mit einer kombinierten (konkomitanten und adjuvanten) Radiochemotherapie mit 6-12 Zyklen Temozolomid.
Bei IDH-mutierten Astrozytomen WHO Grad 3 unterstützen die Ergebnisse der EORTC 26053-22054 CATNON-Studie den Einsatz einer sequenziellen Radiochemotherapie mit 59.4 Gy gefolgt von Temozolomid über 12 Zyklen. Hier ist der Nutzen einer konkomitanten Temozolomid-Therapie parallel zur Radiotherapie noch nicht abschliessend geklärt (7, 8).
Astrozytome WHO-Grad 4, IDH-mutiert sollten wie Glioblastome behandelt werden (Abb. 2). Aufgrund der IDH-Mutation kann von einer Empfindlichkeit gegenüber alkylierenden Substanzen ausgegangen werden.
Neue Therapieansätze für IDH-mutierte Gliome
Neben der Verbesserung des Therapiealgorithmus konventioneller Therapien zielen neue Ansätze auf die IDH-Mutation selbst ab. In den letzten Jahren wurde eine ganze Reihe von IDH-Inhibitoren entwickelt.
Sicherheit, gute Verträglichkeit und biologische Wirkung gemessen an der Reduktion der 2-HG-Produktion im Tumorgewebe konnte in Phase-I-Studien nachgewiesen werden (9). Die bisherigen Effektivitätsdaten mit Tumorvolumenreduktion v. a. bei Patienten mit nicht-kontrastmittelaufnehmenden Tumoren sind ermutigend. Mit der laufenden INDIGO-Studie ist eine internationale randomisierte Phase-III-Studie initiiert, welche die Effektivität (gemessen am PFS) des IDH-Inhibitors AG-881 bei Patienten mit postoperativem Resttumor oder bei progredientem IDH-mutiertem WHO-Grad-2-Gliom untersucht (NCT04164901). Diese Studie rekrutiert Patienten an mehreren Zentren in der Schweiz. Neben spezifischen IDH-Inhibitoren eröffnen spezifische metabolische Abhängigkeiten Möglichkeiten für weitere zielgerichtete Therapien wie beispielsweise DNA-Demethylierungsstrategien oder Glutaminase-Inhibitoren, die bereits in frühen klinischen Studien getestet werden.
Spezifische Impfungen zielen auf die IDH1R132H-Mutation. Die mutierte Sequenz induziert spezifische T-Helferzellen, die das Wachstum IDH-mutierter Tumoren in Tiermodellen kontrollieren können. Die multizentrische NOA-16-Studie (NCT02454634) hat bei Patienten mit neu diagnostizierten IDH1R132H-mutierten Grad 3- und 4-Astrozytomen die Sicherheit und Immunogenität einer IDH1R132H-Peptidvakzine nachweisen können. Die hohe Frequenz an Pseudoprogressionen als Hinweis auf eine intratumorale Immunantwort sowie der Nachweis IDH1R132H-spezifischer T-Helferzellen im Tumorgewebe nach Vakzinierung legen eine biologische Effektivität nahe. Basierend auf präklinischen Hinweisen untersucht die aktuell rekrutierende multizentrische NOA-21-Studie in einer Phase-I-Studie die Sicherheit einer Kombination der IDH1R132H-Vakzine mit dem Immuncheckpoint-Inhibitor Avelumab (NCT03893903). Dabei werden auch mögliche Resistenzmechanismen am Tumorgewebe unter Therapie untersucht (Abb. 1).
Therapie WHO-Grad 1-4, IDH-wild-type-Gliome
Pilozytische Astrozytome WHO-Grad 1, IDH-wild-type
Hintergrund: Mutationen im MAPK/ERK Signalweg wurden in ungefähr 95% pilozytischer Astrozytome (PA) gefunden und gelten mittlerweile als krankheitsdefinierend. Die häufigste molekulare Alteration ist die KIAA1549–BRAF Fusion, welche in 60-70% der Fälle auftritt und mit einer besseren Prognose einhergeht, verglichen mit PA ohne diese Fusion. Die BRAF-V600E-Mutation wird in ca. 10% der PA nachgewiesen.
Therapie: Die initiale Therapie der PA besteht in der maximal möglichen Resektion. Erst bei Tumorprogress oder nicht möglicher Operabilität wird eine Radiotherapie empfohlen und es können auf experimenteller Basis zielgerichtete Therapien eingesetzt werden.
Diffuse Astrozytome WHO-Grad 2 und 3, IDH-wild-type
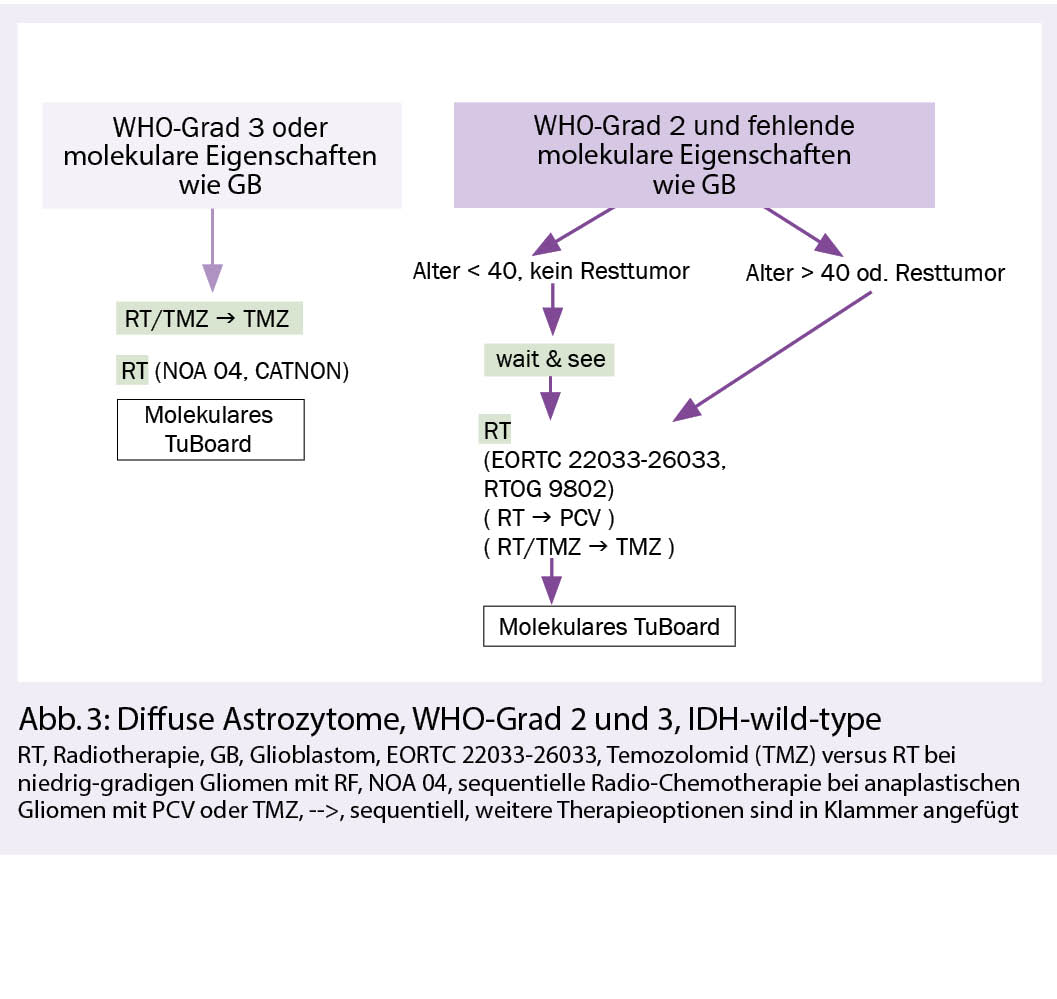
Hintergrund: Diffuse Astozytome IDH-wild-type sind eine heterogene Tumorgruppe, meist mit molekularen Eigenschaften wie bei Glioblastomen und seltener mit einem eher indolentem Verlauf. Bei Gliomen WHO-Grad 2 oder 3, die keine IDH-Mutation aufweisen, kann molekular ein Glioblastom diagnostiziert werden, wenn entweder eine EGFR-Amplifikation oder ein chromosomaler Gewinn auf Chromosom 7 bei komplettem Verlust von einem Chromosom 10 oder aber eine TERT (Telomerase reverse Transkriptase)-Promotor-Mutation vorliegt.
Die postoperative Radiotherapie ist vorläufig die Standardtherapie für Tumoren, die nicht die Kriterien eines Glioblastoms erfüllen. Ein Überlebensvorteil einer zusätzlichen Chemotherapie zur Radiotherapie bei WHO-Grad-2-Astrozytomen war in der RTOG-9802-Studie nur bei Patienten mit IDH-mutierten Tumoren nachweisbar, nicht aber bei IDH-wild-type-Tumoren (3). Es bleibt zu erforschen, weshalb Glioblastome von einer konkomitierenden und adjuvanten Chemotherapie profitieren und Astrozytome vom IDH-wild-type nicht. Die Rolle des MGMT-Promoter-Methylierungsstatus ist bei diesen Gliomen im Gegensatz zu den Glioblastomen auch weniger gut definiert. Nach Versagen der Primärtherapie ist eine molekulare Charakterisierung bei IDH-wild-type-Gliomen mit der Suche therapierbarer Targets eine Option.
Therapie: Nach maximal und neurologisch sicherer Resektion kann in Ausnahmefällen und bei fehlenden klinisch-radiologischen Risikofaktoren (d.h. Alter < 40, kein Resttumor) und WHO-Grad 2 eine initiale Wait-and-see-Strategie verfolgt werden. Bei allen anderen WHO-Grad-2-Astrozytomen empfiehlt sich eine postoperative Radiotherapie mit 50.4-54 Gy optional gefolgt von PCV oder Temozolomid. Bei WHO-Grad 3, IDH-wild-type-Astrozytomen ist aufgrund fehlender Alternativen eine postoperative Therapie analog den Glioblastomen empfohlen, d.h. eine kombinierte Radio-Chemotherapie (59.4-60 Gy) und Temozolomid, gefolgt von einer Temozolomid-Erhaltungstherapie über 6 Zyklen. Bei Kontraindikationen für eine Chemotherapie kann auch eine alleinige RT mit einem Aequivalent von 60 Gy durchgeführt werden (Abb. 3).
Glioblastome WHO-Grad 4, IDH-wild-type, H3-Mutation wild-type
Die Gruppe der Glioblastome enthält auch histologische Sub-typen wie das Riesenzell-Glioblastom, das Gliosarkom und epitheloide Glioblastome (welche in bis zu 50% eine BRAF-Mutation aufweisen).
Therapie: Die Therapierichtlinien berücksichtigen das Alter, den Karnofsky-Performance-Status (KPS) und den MGMT-Promoter-Methylierungsstatus, wie im Algorithmus in Abb. 4 dargestellt.
Die postoperative Therapie bei Glioblastomen wurde 2005 weltweit standardisiert mit konkomitierender Radio-Chemotherapie mit 60 Gy und Temozolomid, gefolgt von einer Erhaltungstherapie mit Temozolomid über 6 Zyklen (10). 2019 wurde die NOA-09-Studie publiziert, welche zusätzlich zur Standardtherapie bei methyliertem MGMT-Promoter Lomustin (CCNU) ergänzt (11). Die Kombination beider Alkylantien, Temozolomid und Lomustin, hat bei insgesamt kleiner Patientenzahl zwar zu einer Verlängerung des OS, aber auch zu erhöhter Knochenmarkstoxizität geführt, weshalb diese Chemotherapie-Kombination eher bei jungen Patienten und nicht breit Anwendung findet. Für neu diagnostizierte Glioblastome ist optional die Tumor-Treating-Field-Therapie (TTFields) in der Schweiz und in anderen europäischen Ländern zugelassen. Es handelt sich dabei um elektrische Wechselfelder mit niedriger Intensität und mittlerer Frequenz (200 kHz), die über der Tumorregion mit speziellen Transducern angelegt werden. Diese Therapie, welche der Patient über mindestens 18 Stunden pro Tag tragen soll, hat in einer randomisierten Phase-III-Studie zu einer Verlängerung des PFS und des OS geführt verglichen mit der Standardtherapie bei Glioblastom. TTFields werden, bis auf mögliche Hautirritationen, gut vertragen und sind in ihrer Wirkung unabhängig von Glioblastom-Subgruppen (12). Bei hohem Alter und schlechtem KPS ist im Gespräch mit Patient und seinen Angehörigen auch «best supportive care» eine mögliche gemeinsame Entscheidung.
Bevacizumab, als hauptsächlich Ödem-reduzierende, antiangiogene Substanz, hat bei neu diagnostizierten Glioblastomen in zwei grossen randomisierten Studien keinen Überlebensvorteil gebracht und wird deshalb nur im Rezidiv oder bei Radionekrose zur Symptombeeinflussung eingesetzt.
Es gibt bislang noch keine positiven Studiendaten, welche einen Immuncheckpoint-Inhibitor in der postoperativen Phase zusätzlich zur Standardtherapie stützen. Die Suche nach Subgruppen, für welche eine Immuntherapie einen Vorteil bringen könnte, geht weiter. Studien hierzu sind in der Schweiz offen und können bei den Verfassern dieses Artikels angefordert werden.
Histon-mutierte Gliome, IDH-wild-type
Hintergrund: Histon H3.3 Missense-Mutationen beeinflussen die epigenetische Regulation der Genexpression.
Therapie: Diffuse Mittelliniengliome mit der Mutation H3K27M, WHO-Grad 4 werden mit 54-60Gy radiotherapiert, alternativ chemo-radiotherapiert mit Temozolomid analog den Glioblastomen, aber noch mit ungenügender Evidenzlage.
Diffuse Hemisphären-Gliome mit der Mutation H3.3.G34, WHO-Grad 4 sind zu einem hohen Prozentsatz MGMT methyliert und werden chemo-radiotherapiert mit Temozolomid analog den Glioblastomen. Bisher liegen für diese vergleichsweise neuen Tumorentitäten nur limitierte Daten und Erfahrungswerte vor.
Das diagnostische und therapeutische Management bei Patienten mit neu diagnostizierten Gliomen sollte den Empfehlungen eines interdisziplinären neuro-onkologischen Tumorboards folgen, welche sich auf die neue WHO-Klassifikation, die aktuelle Datenlage abgeschlossener Studien, den Zugang zu laufenden Studien oder auf einen internationalen Konsensus abstützen.
Dabei sind der Allgemeinzustand und die Bedürfnisse der Betroffenen zu berücksichtigen. Neurorehabilitation, Psychoonkologie und soziale Beratung gehören zur ganzheitlichen Betreuung. Für weiterführende Informationen verweisen wir auf die aktuelle EANO (Europiean Association of Neuro-Oncology) Guideline (13).
Copyright bei Aerzteverlag medinfo AG
Universitätsspital Zürich
Institut für Pathologie und Molekularpathologie
Schmelzbergstrasse 12
8091 Zürich
silvia.hofer@usz.ch
Institut für Radio-Onkologie
Kantonsspital Graubünden
Loëstrasse 170
7000 Chur
Leitender Arzt, Klinik für Neurologie
Universitätsspital Zürich
Frauenklinikstrasse 26
8091 Zürich
Die Autorinnen und der Autor haben in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
1. Friedrich M, Bunse L, Wick W et al.: Perspectives of Immunotherapy in isocitrate dehydrogenase-mutant gliomas. Curr Opin Oncol 30(6): 368-374, 2018. DOI: 10.1097/CCO.0000000000000478
2. Buckner JC, Chakravarti A, Curran WJ Jr et al.: Radiation plus procarbazine, CCNU, and vincristine in Low-Grade Glioma. NEJM 374:1344-1355, 2016. DOI:10.1056/NEJMc1605897
3. Bell EH , Zhang P, Shaw EG et al.: Comprehenisve Genomic Analysis in NRG
Oncology/RTOG 9802: A Phase III Trial of Radiation versus Radiation plus Procarbazine, Lomustine (CCNU), and Vincristine in High-Risk Low Grade Glioma. J Clin Oncol 38:3407-3417, 2020. DOI: 10.1200/JCO.19.02983
4. Baumert BG, Hegi ME, van den Bent MJ et al.: Temozolomide chemotherapy
versus radio- therapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 17(11):1521-1532, 2016. DOI:10.1016/ S1470-2045(16)30313-8
5. Cairncross G, Wang M, Shaw E et al.: Phase III trial of chemo- radiotherapy for anaplastic oligodendroglioma: long-term re- sults of RTOG 9402. J Clin Oncol 31: 337–343, 2013. DOI:10.1200/JCO.2012.43.2674
6. van den Bent MJ, Brandes AA, Taphoorn MJ, et al.: Adjuvant procarbazine,
lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol 31: 344–350, 2013. DOI: 10.1200/JCO.2012.43.2229
7. van den Bent MJ, Baumert B, Erridge SC et al.: Interim results from the CATNON trial (EORTC study 26053-22054) of treatment with concurrent and adjuvant temozolomide for 1p/19q non-co-deleted anaplastic glioma: a phase 3, randomised, open-label intergroup study. Lancet 390(10103):1645-1653, 2017. DOI:10.1016/S0140-6736(17)31442-3. Erratum in: Lancet. 390(10103):1644, 2017
8. van den Bent MJ, Tesileanu CMS, Wick W et al.: 2nd interim analysis and IDH status of the EORTC randomized phase III CATNON trial on adjuvant and concurrent temozolomide in anaplastic astrocytoma. Lancet Oncology 22 (6) 813-823, 2021. DOI:https://doi.org/10.1016/S1470-2045(21)00090-5
9. Mellinghoff I, Ellingson BM, Touat M et al.: Ivosidenib in Isocitrate Dehydrogenase 1 Mutated Advanced Glioma. J Clin Oncol 38(29): 3398-3406, 2020. DOI: 10.1200/JCO.19.03327
10. Stupp R, Mason WP, van den Bent MJ et al.: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996, 2005. DOI:10.1056/ NEJMoa043330
11. Herrlinger U, Tzaridis T, Mack F, et al.: Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): a randomised, open-label, phase 3 trial. Lancet 393(10172):678-688, 2019. DOI:10.1016/S0140-6736(18)31791-4
12. Stupp R, Tailllibert S, Kanner A et al.: Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma.A Randomized Clinical Trial. JAMA 318 (23): 2306-2316, 2017. DOI:10.1001/jama.2017.18718
13. Weller M, van den Bent M, Preusser M et al.: EANO Guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nature Reviews | Clinical Oncology 2020. doi.org/10.1038/s41571-020-00447-z



info@onco-suisse
- Vol. 11
- Ausgabe 4
- August 2021