
Die konstitutionelle klinische Blutungsneigung ist Realität, diese beruht sowohl auf erworbenen wie auch auf hereditären Veränderungen der Gerinnung. Junge Menschen mit weniger Expositionsrisiken im Vergleich zu den Älteren fallen klinisch nicht sofort auf, deshalb ist eine gründliche, frühe Labordiagnostik sehr hilfreich und sinnvoll.
Die hämostatischen Mechanismen sind ein komplexes biologisches System, wo verschiedene lösliche Gerinnungsenzyme in Interaktion mit Zellstrukturen eine kaskadenartige, gegenseitige Aktivierung abspielen. Das System bleibt «eingefangen» im Gefässnetz und befindet sich primär in Ruhezustand, bis eine mechanische oder toxische Verletzung der Gefässwand vorkommt. Die Verletzung bedeutet Blutverlust und ist gleichzeitig der Trigger für den Start der Reaktionskaskade, welche unausweichlich in die Bildung des hämostatischen Thrombus führt. Die Reaktionspartner sind heute bekannt, sowohl in ihrer Struktur wie auch in ihrer Funktion (Abb. 1).
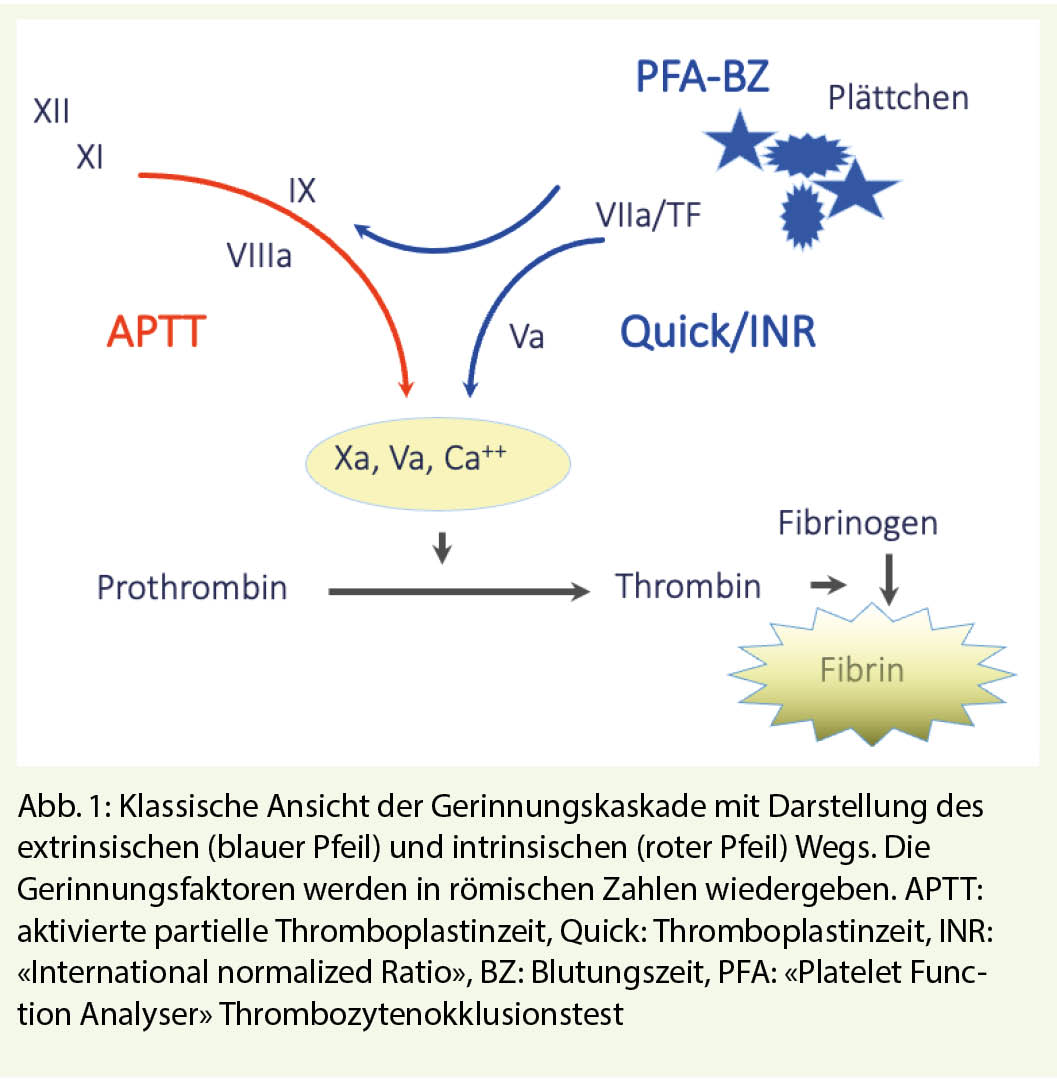
Eine Abweichung aus diesem physiologischen Weg, im Sinne eines Mangels eines Gerinnungsfaktors oder einer Hemmung dessen Funktion, bedeutet Verlangsamung der Gerinnung, klinisch als Blutungsneigung erkannt.
Zeichen der Blutungsneigung sind die Petechien (stecknadelkopfgrosse Flecken), die Ekchymosen (münzengrosse Flecken), die Suffusionen (grossflächige diffuse Blutungen) oder die Hämatome (abgrenzbare Blutungen).
Aus der Art der Blutung kann man nur bedingt die Ursache extrapolieren. Vaskuläre Defekte äussern sich meistens als Hämatome, Hautblutungen oder gastrointestinale Blutungen. Thrombozytäre Defekte lösen Petechien, Zahnfleischblutungen, Nasenblutungen, Gastrointestinal- oder ZNS-Blutungen aus. Störungen der plasmatischen Gerinnung manifestieren sich oft als Hämatome in den Muskellogen, als Hautblutungen, intraartikuläre Blutungen oder Blutungen nach Trauma.
Eine sorgfältige Anamnese, die klinische Untersuchung und die gezielte Laborabklärung führen fast immer zur genauen Identifizierung der Gerinnungsstörung.
Erste Abklärung
Anamnese
Eine gezielte detaillierte Blutungsanamnese ist sehr hilfreich. Da die Patienten die vereinzelten früheren Blutungsepisoden oft vergessen oder verdrängen, sollten sie vor blutigen diagnostischen und therapeutischen Eingriffen gezielt befragt werden (Tabelle 1). Strukturierte Fragebögen im Sinne eines Scores, wie der Blutungserfassungsscore BAT-ISTH“ der Internationalen Gesellschaft für Thrombose und Hämostase ISTH, können auch hilfreich sein. Starke Menstrualblutungen seit der Menarche erfordern Abklärung auf von Willebrand-Krankheit. Je niedriger der von Willebrand Faktor, desto stärker ist die Blutungsneigung.

Objektive Befunde
Bei den milden Gerinnungsstörungen kommt es zu einem Befund erst bei Provokationen (Trauma, Operation, Antikoagulation), bei den mittelschweren oder schweren Defekten können Blutungen auch spontan auftreten. Bei älteren schweren Hämophilen liegen eindrückliche Störungen des Bewegungsapparates (hämophile Arthropathie, Muskel-atrophie, Kontrakturen, Deformitäten etc.) vor, bei jüngeren hingegen fehlen in der Regel wegen der prophylaktischen Substitution solche Residuen. Neben eindeutigen akuten Blutungsmanifestationen müssen auch unklare Beschwerden bei hereditärer Blutungsneigung bis zum Beweis des Gegenteils primär als blutungsbedingt angesehen werden.
Labordiagnostik
Bei Verdacht auf hereditäre oder erworbene Blutungsneigung soll eine Basisdiagnostik verordnet werden. Diese beinhaltet ein Blutbild, die Globaltests der Gerinnung und der Plättchenfunktion (Thrombozytenokklusionstest PFA). Ergänzend werden auch der von Willebrand Faktor und Faktor XIII verlangt, weil diese durch die Globaltests nicht erkannt werden (Tabelle 2). Damit erfasst man die häufigen und seltenen Störungen der Hämostase. Je nach Resultat der Basisdiagnostik wird die Abklärung mit Spezialanalysen erweitert, wie z.B. gezielte Bestimmung der Gerinnungsfaktoren oder Analyse der Thrombozytenfunktion mittels Plättchenaggregation und/oder Immunphänotypisierung (Tabelle 3).


Spezialanalytik
Gezielte Untersuchung der Plättchenaggregation
Die Plättchenaggregation untersucht und klassifiziert Störungen der Plättchenfunktion, und liefert Hinweise auf Defekte der Plättchenmembranrezeptoren. Dabei wird in vitro die Stimulierbarkeit der Plättchen durch verschiedene natürliche Agonisten (ADP, Kollagen, Adrenalin, Ristocetin, Arachidonsäure, Thrombin (TRAP) und Thromboxanrezeptor-Agonist) turbidimetrisch bestimmt.
Gezielte Untersuchung der Plättchenmembranrezeptoren (Immunphänotypisierung)
Diese werden bei V.a. hereditäre Thrombopathien nachbestellt und helfen zur Diagnose und Typisierung von Defekten der Plättchenmembranrezeptoren. Dabei wird mit monoklonalen Antikörpern und Durchflusszytometrie die Rezeptorendichte auf der Plättchenoberfläche vor und nach in-vitro Plättchenaktivierung semi-quantitativ bestimmt.
Von Willebrand Faktor-Diagnostik und Multimere (VWF-MM)
Das von Willebrand Syndrom ist die häufigste hereditäre Blutungsneigung (Inzidenz 1/200-300) und wird durch die Bestimmung des von Willebrand Faktors (Funktion und Antigen) sowie des Faktors VIII erfasst. Die Abklärung wird durch die Analyse der VWF-MM ergänzt. Diese wird bei V.a. von Willebrand-Syndrom jeden Typs nachbestellt und hilft zur Diagnose und Typisierung des vWF-Defektes. Die Methode besteht in der qualitativen und quantitativen Charakterisierung der zirkulierenden vWF-Multimere mittels Gel-Elektrophorese.
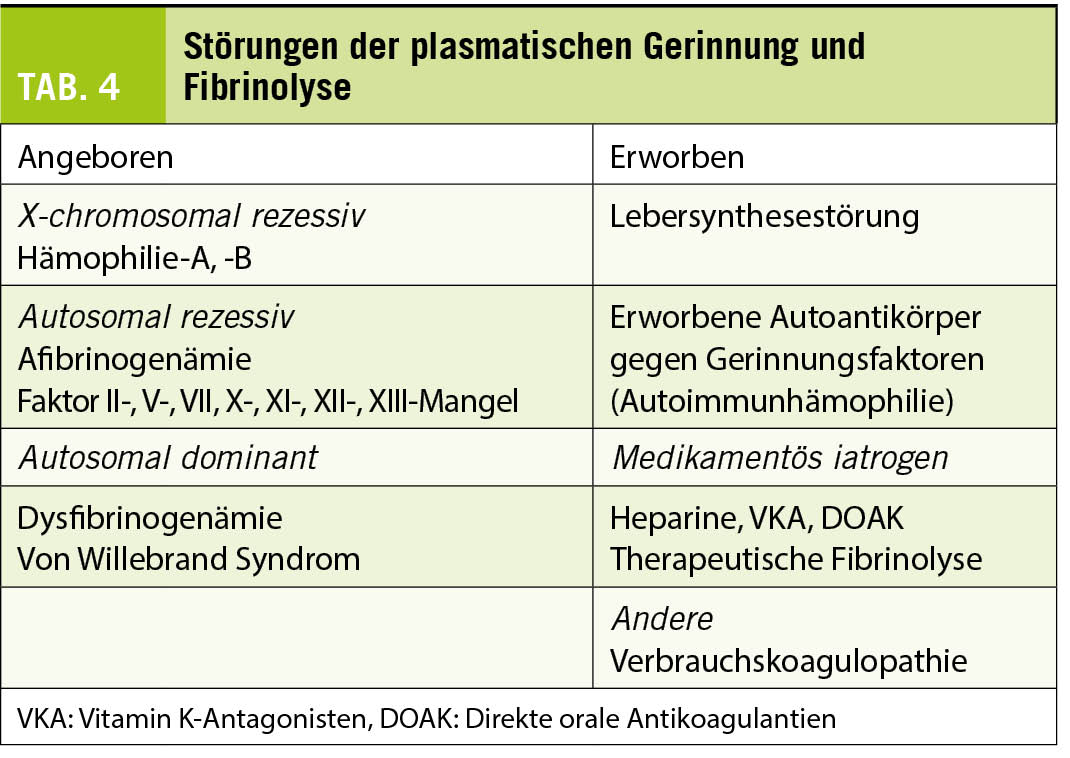
Klinik und Diagnostik der Blutungsneigung (Tabelle 4)
Hämophilie A und B
Die Hämophilie ist eine X-chromosomal vererbte Verminderung der Gerinnungsfaktoren VIII oder IX, Männer sind betroffen, Frauen sind Konduktorinnen. Klinisch sind Gelenksblutungen und Muskelblutungen typisch. Bei Trauma oder Operation ist aber mit ebenso massiven Blutungen zu rechnen.
Bei schwerer und mittelschwerer Hämophilie A oder B ist die APTT eindeutig verlängert, bei milder Hämophilie ist sie nur grenzwertig erhöht oder noch im Normbereich. Eine normale APTT erlaubt daher den Ausschluss einer milden Hämophilie nicht, wenn ein Verdacht aufgrund der Anamnese vorliegt. Die gezielte Bestimmung des Faktors VIII oder IX identifiziert genau den Defekt.
Von Willebrand-Syndrom
Autosomal vererbte Störung des von Willebrand-Faktors (vWF) als Folge einer Vielzahl von Abnormitäten des von vWF-Gens. Der von Willebrand-Faktor ist wichtig für die Adhäsion der Plättchen an die verletzte Gefässintima und ihre Aktivierung. Zudem stabilisiert er den Faktor VIII, mit dem er im Plasma komplex-gebunden zirkuliert. Zahlenmässig überwiegen die milden Formen, die nur bei Provokationen bluten und meistens eine normale Blutungszeit aufweisen. Alle Typen zusammengenommen sollen nach gewissen Schätzungen fast 1% der Bevölkerung betreffen. Die schwerste Form (Typ 3) hat eine Inzidenz von 1 pro Mio.
Die Diagnostik beruht auf der quantitativen Bestimmung des von Willebrand-Faktors (Funktion und Antigen) und der Faktor VIII-Aktivität. Die Typeneinteilung ist dauernd im Fluss, wobei eine Klassierung nach einigen Phänotypen den klinischen Bedürfnissen bisher genügt.
Typ 1: Konkordante Verminderung von funktionellem und immunologisch gemessenem vWF auf weniger als 50% der Norm (meistens 5-30%). Faktor VIII um 50% oder tiefer. Autosomal dominant, macht etwa 70% der Patienten aus. Cave: Menschen mit Blutgruppe O haben physiologisch tiefere vWF-Werte ohne klinische Blutungsneigung. Der Cut-Off wird hier auf 35% statt 50% gesetzt.
Typ 2A: Verminderung der vWF-Aktivität begleitet von normalem oder nur leicht vermindertem vWF-Antigen (Ratio vWF Aktivität/vWF Antigen <0.7). Die Analyse der Multimere ergibt eine Verminderung der funktionell besonders aktiven hochmolekularen Ketten. Faktor VIII normal oder grenzwertig reduziert. Autosomal dominante Vererbung (einige rezessiv).
Typ 2B: Wie Typ 2A, jedoch begleitet von einer paradoxen Verstärkung der Ristocetin-induzierten Aggregation des plättchenreichen Patientenplasmas, bedingt durch eine erhöhte Affinität abnormer vWF-Ketten für den GP Ib/V/IX-Rezeptor des Patienten (von Willebrand Faktor Rezeptor). Bei der vWF-Multimeren Analyse Verminderung der grossen und mittelgrossen VWF-Ketten. Stimulation der Freisetzung des abnormen vWF führt zur Thrombopenie. Autosomal dominante Vererbung.
Typ 2M: Schlechte Plättchenaffinität der vWF-Multimere. Im Unterschied zu den Typen 2A und 2B sind auch die grossen Multimere vorhanden, aber funktionsgestört, was sich in der erniedrigten vWF-Funktion bei noch normalem vWF-Antigen und normaler vWF-Multimerenelektrophorese äussert (jedoch abnorme Form der Banden-Tripletten der Multimere). Autosomal dominante Vererbung.
Typ 2N, vWF-Typ Normandy: vWF mit abnormer Bindungsstelle für den Faktor VIII, der wegen fehlender Bindung an den sonst quantitativ und qualitativ normalen vWF schnell eliminiert wird. Formal entsteht das Bild einer leichten Hämophilie A (auch bei Frauen) mit VIII um 5-30%. Autosomal rezessive Vererbung.
Typ 3: vWF funktionell und immunologisch nicht nachweisbar. Faktor VIII wegen fehlendem Transportprotein im Plasma auch auf wenige Aktivitätsprozente reduziert. Autosomal rezessive Vererbung. Blutungszeit stark verlängert.
Seltene Koagulopathien
Die sehr seltenen hereditären Verminderungen einzelner Faktoren äussern sich in abnormen Globaltests: II, V und X durch abnorme Quick/INR und abnorme APTT, VII durch abnorme Quick/INR bei normaler APTT und die Kontaktphasenproteine XII und Faktor XI durch isolierte Verlängerung der APTT. Bei Afibrinogenämie fehlt die Gerinnselbildung in allen Globaltests. Die Diagnose wird mit der spezifischen Faktorenbestimmung gestellt. Der Mangel an Faktor XII ist nicht mit einer Blutungsneigung verbunden, ebensowenig der Faktor VII-Mangel mit Restaktivitäten über 10% der Norm.
Hereditäre Thrombozytenfunktionsstörungen
Die schwersten Blutungen findet man bei den seltenen, klassischen Modellkrankheiten der Plättchen, der Thrombasthenie Glanzmann (Defekt des Fibrinogenrezeptors GP IIb/IIIa) und dem Bernard-Soulier-Syndrom (Defekt des vWF-Rezeptors GP Ib/V/IX). Die Blutungszeit ist in der Regel abnorm, spezifische Diagnostik mit Plättchenaggregationstests und Rezeptorenuntersuchung bestätigt den Defekt. Weitere, unterschiedlich charakterisierte Defekte betreffen andere Oberflächenrezeptoren, die Signalübertragung, die Alpha- und Dichte-Granula sowie den Arachidonsäuremetabolismus.
Dr. med. Leda Leoncini1
Dr. med. Mario Uhr1
Dr. Yordanka Tirefort2
Prof. Dr. med. Dimitrios Tsakiris3
1 SYNLAB Suisse SA, Via Pianon 7, 6934 Bioggio
(leda.leoncini@synlab.com, mario.uhr@synlab.com)
2 SYNLAB Suisse SA, Ch. d’Entre-Bois 21, 1018 Lausanne
(yordanka.tirefort@synlab.com)
3 SYNLAB Suisse SA, Alpenquai 14, 6002 Luzern
(dimitrios.tsakiris@synlab.com)
Copyright Aerzteverlag medinfo
SYNLAB Suisse SA
Via Pianon 7
6934 Bioggio
leda.leoncini@synlab.com
Klinik für Hämatologie
Hämatologische Diagnostik Labormedizin
Universitätsspital Basel und Blutspendezentrum beider Basel SRK
Petersgraben 4
4031 Basel
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel zu deklarieren.
1. Hayward CPM. How I investigate for bleeding disorders. Int J Lab Hematol. 2018 May;40 Suppl 1:6-14. doi: 10.1111/ijlh.12822. PMID: 29741250.
2. Boender J, Kruip MJ, Leebeek FW. A diagnostic approach to mild bleeding disorders. J Thromb Haemost. 2016 Aug;14(8):1507-16. doi: 10.1111/jth.13368. Epub 2016 Jun 27. PMID: 27208505.
3. Hayward CPM, Moffat KA, Brunet J, Carlino SA, Plumhoff E, Meijer P, Zehnder JL. Update on diagnostic testing for platelet function disorders: What is practical and useful? Int J Lab Hematol. 2019 May;41 Suppl 1:26-32. doi: 10.1111/ijlh.12995. PMID: 31069975.
4. James PD. Women and bleeding disorders: diagnostic challenges. Hematology Am Soc Hematol Educ Program. 2020 Dec 4;2020(1):547-552. doi: 10.1182/hematology.2020000140. PMID: 33275722; PMCID: PMC7727580.
5. Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, Neunert C, Lillicrap D; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010 Sep;8(9):2063-5. doi: 10.1111/j.1538-7836.2010.03975.x. PMID: 20626619


der informierte @rzt
- Vol. 11
- Ausgabe 12
- Dezember 2021