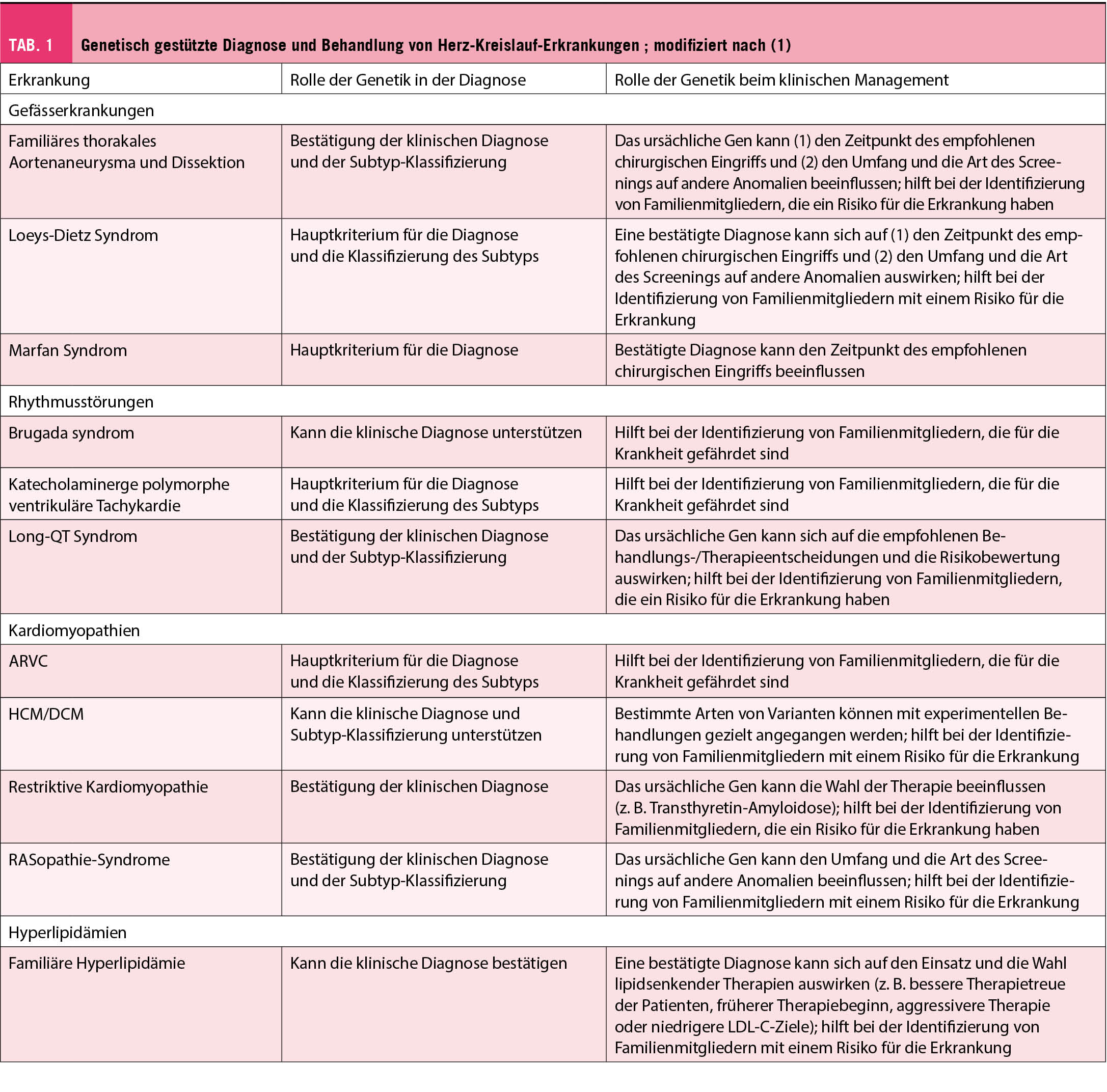
Viele kardiologische Leitlinien und Guidelines beziehen genetische Daten in die Empfehlungen für die Diagnose und das personalisierte klinische Management ein. Daher sind die Grundprinzipien der kardiovaskulären Genetik für klinische Kardiologen hochrelevant. Sie müssen erkennen, bei welchen Patienten eine genetische Grunderkrankung vorliegen könnte und diese gegebenenfalls zur genetischen Testung überweisen. In diesem Übersichtsartikel werden die Grundprinzipien der medizinischen Genetik zusammengefasst. Ein besonderer Fokus liegt auf einem praktischen Ansatz für klinische Gentests konzentriert auf die folgenden drei Kategorien von Herz-Kreislauf-Erkrankungen: Aortopathien, Kardiomyopathien und Herzrhythmusstörungen.
Abstract: Many cardiology guidelines incorporate genetic data into recommendations for diagnosis and personalized clinical management. Therefore, the basic principles of cardiovascular genetics are highly relevant to clinical cardiologists. They need to identify which patients may have an underlying genetic disease and refer them for genetic testing when appropriate. This review article summarizes the basic principles of medical genetics. A particular focus is on an approach to clinical genetic testing focused on the following three categories of cardiovascular disease: aortopathiy, cardiomyopathies, and cardiac Arrhythmias.
Key Words: cardiogenetics, cardiac arrhythmia, cardiomyopathy, aortopathy
In den letzten zehn Jahren wurden zunehmend genetische Ursachen für viele Arten von Herz-Kreislauf-Erkrankungen erkannt. Dies hat je nach genetischer Erkrankung erhebliche Auswirkungen auf die Behandlung der Patienten.
Genetische Diagnosen waren früher schwierig. Die Technik der massiv parallelen DNA-Sequenzierung, sowie kommerzielle Grosslabors haben Kosten- und Verfügbarkeitsproblematik von genetischen Tests, Gen-Panels und gar Exom- und Genom-Sequenzierung aber beseitigt. Dutzende bis hunderte von Genen, die von Interesse sind, können heute zu geringeren Kosten und viel schneller sequenziert werden, so dass klinische Tests leichter zugänglich sind als je zuvor.
Wen soll man testen?
Ein zentraler Grundsatz der klinischen Genetik besteht darin, bei einem eindeutig betroffenen Patienten (dem sogenannten Indexfall) mit der genetischen Untersuchung zu beginnen. So wird die Wahrscheinlichkeit der Identifizierung einer pathologischen Variante (PV) maximiert. Wenn mehr als ein Familienmitglied betroffen ist, sollte in der Regel derjenige Patient untersucht werden, bei dem die Krankheit in einem jüngeren Alter ausgebrochen ist oder der keine störenden Umweltfaktoren aufweist, die zu einem ähnlichen klinischen Bild führen könnten. Wenn der Indexfall bei der Autopsie identifiziert wird, kann eine postmortale Blut- oder Gewebeprobe für die DNA-Analyse entnommen werden.
Steht kein betroffener Patient für die Untersuchung zur Verfügung, kann eine kardiale Phänotypisierung von Verwandten ersten Grades mit Bildgebung, EKG, Belastungstests usw. sinnvoll sein, wenn es eine Familienanamnese für eine potenziell vererbte kardiogenetische Erkrankung gibt.
Wird durch Gentests eine PV identifiziert, die zu den klinischen Merkmalen passt, wird eine Kaskadenuntersuchung von Familienmitgliedern empfohlen. Durch die Untersuchung beider Elternteile kann die risikobehaftete Seite der Familie identifiziert werden. Wenn beide Elternteile negativ getestet werden, könnte die PV beim Probanden de novo aufgetreten sein. Ein sogenannter Mosaizismus in einem nicht betroffenen Elternteil kann zu mehreren betroffenen Nachkommen führen. Deshalb werden Gentests für Geschwister angeboten, auch wenn die Tests der Eltern negativ sind.
Jedes Kind eines Patienten mit einer autosomal dominant vererbten Herzerkrankung hat ein 50%iges Risiko, das pathogene Allel geerbt zu haben, und ein 50%iges Risiko, das normale Allel geerbt zu haben. Ein Geschwisterkind oder ein Kind, das negativ auf eine bekannte familiäre PV getestet wurde, ist nicht gefährdet, die Krankheit zu entwickeln, muss nicht weiter untersucht werden und kann die Krankheit nicht an Kinder weitergeben.
Fallbeispiel 1: Long-QT Syndrom
22-jährige Patientin wird wegen rezidivierenden epileptischen Anfällen trotz antikonvulsiver Medikation in einer neurologischen Abteilung stationär abgeklärt. Während die Patientin im Rahmen der Routinediagnostik ein Langzeit-EKG trägt, erleidet sie erneut einen epileptischen Anfall. Nachträglich ergibt die Auswertung des Langzeit-EKG eine Torsade-des-Pointes-Tachykardie zum Zeitpunkt des vermeintlichen epileptischen Anfalls, die spontan terminiert. Daraufhin fällt erstmalig im Ruhe-EKG eine verlängerte QTc-Zeit von 485 ms auf. Es wird die Diagnose eines LQTS gestellt und es erfolgt die Implantation eines Defibrillators. Nachbetreuende Kardiologe überweist die Patientin im Verlauf in eine Humangenetik, wo der Nachweis einer KCNH2-Mutation und somit eines LQT 2 erfolgt.
Daraufhin folgt nochmals eine gezielte Anamneseerhebung, in der die Patientin erstmalig von durch Schulterklopfen oder Erschrecken ausgelöste Synkopen im Teenageralter berichtet. Die Patientin kann daraufhin über die für das LQT 2 typischen Auslösemechanismen aufgeklärt werden.
Fallbeispiel 2: Arrhythmogene rechtsventrikuläre Kardiomyopathie
In einer Familie mit 3 Söhnen traten beim ältesten Sohn im Alter von 17 Jahren wiederholte Synkopen im Rahmen von ventrikulären Tachykardien auf. Die durchgeführten bildgebenden Untersuchungen ergaben dabei keinen Hinweis auf eine strukturelle Herzerkrankung.
Unter der Annahme einer idiopathischen rechtsventrikulären Ausflusstrakt-Tachykardie wurden mehrfach Versuche einer interventionellen Ablation vorgenommen. Diese waren erfolglos, sodass dem Patienten ein Defibrillator implantiert wurde. Nach Implantation traten gehäuft durch Arrhythmien getriggerte adäquate Schocks auf, die sich wiederum nur schwer mit Antiarrhythmika kontrollieren liessen und so zu einer starken Traumatisierung des Patienten und seiner Familie führten. Bis zum Alter von 30 Jahren stellte sich jedoch eine relative elektrische Ruhe mit seltenen Ereignissen ein. Der zweite Sohn dieser Familie stellte sich im Alter von 24 Jahren mit dem klinischen Bild einer Myokarditis in einer kardiologischen Klinik vor. Während der akuten Phase mit erhöhten kardialen Markern wurde eine ventrikuläre Tachykardie dokumentiert. Die Echokardiografie, rechtsventrikuläre Angiografie und das kardiale MRT ergaben keinen Hinweis auf eine Arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC). Aufgrund der Familienanamnese erfolgte jedoch die Vorstellung in einem tertiären Zentrum zur weiteren Abklärung. Wenige Tage nach Vorstellung erlitt der zweite Sohn einen plötzlichen Herztod. Erst später erfolgte eine molekulargenetische Untersuchung: eine in der Literatur mehrfach beschriebene Mutation im Plakophilin-2-Gen (PKP2) und somit der Nachweis einer ARVC. In der daraufhin erfolgten Durchsicht alter Befunde des ältesten Sohnes fanden sich dann Hinweise auf strukturelle Auffälligkeiten des rechten Ventrikels. Die erneute Ansicht des externen MRT des zweiten Sohnes ergab ebenfalls strukturelle Auffälligkeiten im rechten Ventrikel.
Was soll man testen?
Wenn bei einem Familienmitglied eine PV identifiziert wurde, ist es wichtig, einen weiteren genetischen Bericht zur Bestätigung einzuholen und zu überprüfen. Wurde ein Gen-Panel durchgeführt und ein einzelner PV identifiziert, können die Kaskadentests für Familienmitglieder auf den einzelnen PV beschränkt werden. Wurden keine Gentests durchgeführt oder waren sie nicht diagnostisch, ist eine genetische Untersuchung des Herzens am effizientesten und wirtschaftlichsten, wenn ein Multigen-Panel bestellt wird. Die Gene, die in einem bestimmten Panel enthalten sind (z. B. Kardiomyopathie oder Aortopathie), variieren von Labor zu Labor, die Anzahl der Gene variiert im Laufe der Zeit und es ist zu berücksichtigen, ob der Test eine Deletions-/Duplikationsanalyse umfasst. Bei der Bestellung grosser Panels oder der Exom-Sequenzierung wird die Konsultation eines klinischen Genetikers oder genetischen Beraters empfohlen. Es hat sich gezeigt, dass die Überprüfung genetischer Testbestellungen vor der Testdurchführung durch einen genetischen Berater im Labor den Anteil unangemessener Tests um 26 % reduziert.
Wann sollte getestet werden?
Einige der häufigsten Indikationen für eine genetische Untersuchung des Herzens sind: Kardiomyopathien, plötzlicher Herzstillstand bei jungen Menschen, vererbte Herzrhythmusstörungen, Muskeldystrophien oder Friedreich-Ataxie in Verbindung mit Kardiomyopathie, Aortopathien, angeborene Herzkrankheiten und vererbbare Fettstoffwechselstörungen. Ein jüngeres Erkrankungsalter und eine positive Familienanamnese erhöhen die Wahrscheinlichkeit, eine genetische Ätiologie zu identifizieren. Die Erstellung eines Familienstammbaums über drei Generationen ist ein Standardbestandteil der genetischen Untersuchung und kann wichtige Anhaltspunkte für die Diagnose liefern und aufzeigen welche Familienmitglieder gefährdet sind. Kardiovaskuläre genetische Störungen bei Kindern können autosomal rezessiv, autosomal dominant, X-chromosomal oder mitochondrial vererbt werden. Im Erwachsenenalter auftretende kardiovaskuläre genetische Störungen werden in der Regel autosomal-dominant vererbt.
Phänotypische, kardiale Merkmale
Syndromale genetische Erkrankungen umfassen Merkmale in mehreren Organsystemen und werden häufig in der Kindheit diagnostiziert. Bei Erwachsenen können phänotypische Merkmale, die auf eine genetische Erkrankung hindeuten, auf der Anamnese, der körperlichen Untersuchung oder bildgebenden Befunden beruhen. Zu den Merkmalen einiger Patienten mit Marfan-Syndrom gehören zum Beispiel Hochwuchs, Arachnodaktylie und eine Verlagerung der Augenlinse nach oben. Patienten mit Loeys-Dietz-Syndrom können einen Hypertelorismus und ein bifides Zäpfchen aufweisen. In der Bildgebung ist die Form der Aortensinus und der sinotubulären Kreuzung bei einer genetischen Aortopathie typischerweise ganz anders als bei einer hypertensiven Gefässerkrankung.
Fallbeispiel 3: Hypertrophe obstruktive Kardiomyopathie (HOCM)
40-jähriger Mann mit bekannter familiärer HOCM stellt sich zur genetischen Abklärung der Erkrankung vor. Wenige Jahre zuvor erfolgreiche interventionelle Septumablation mit Alkoholinjektion (TASH: transkoronare Ablation der Septumhypertrophie). Der Sohn des Patienten ist im Alter von 13 Jahren aufgrund eines plötzlichen Herztods verstorben. Zuvor war bei ihm bereits ebenfalls eine HOCM bekannt. Weitere Familienanamnese war bis auf einen unklaren plötzlichen Todesfall im Alter von 33 Jahren bei einem Onkel mütterlicherseits bland. Genetische Diagnostik: Nachweis einer Splice-Mutation im Gen für das myosinbindende Protein C (MYBPC3). Diese Mutation wurde auch bei den beiden Töchtern (22 und 19 Jahre) nachgewiesen, wobei die ältere Tochter im Kardio-MRT grenzwertige Wandstärken aufwies. Beim klinisch asymptomatischen älteren Bruder des Indexpatienten wurde ebenfalls die Mutation nachgewiesen, gleichzeitig fand sich in Echokardiografie und MRI der Nachweis einer nicht obstruktiven HCM.
Fazit: Früher an die Genetik denken
Leider werden viele Patienten mit einer genetischen Erkrankung erst nach einem akuten klinischen Ereignis, wie z. B. einem wiederbelebten Herzstillstand, einer Aortendissektion oder einer akuten Herzinsuffizienz, medizinisch betreut. Andere kardiovaskuläre Ereignisse, die potenziell auf eine genetische Grunderkrankung hindeuten, sind z. B. die Dissektion der Halsschlagader, Synkopen bei körperlicher Anstrengung oder strukturelle Anomalien, die bei bildgebenden Verfahren oder bei der Autopsie festgestellt werden.
Ein wünschenswertes Beispiel dafür, wie sich Gentests auf die Prävention und Behandlung von Herzrhythmusstörungen auswirken könnten:
- Derzeitiges Modell: Patienten werden wegen unspezifischer Beschwerden wie Schwindel, Herzklopfen oder Synkopen von verschiedenen Fachärzten untersucht. Ein Herzstillstand kann der erste Kontakt mit einem Kardiologen sein.
- Zukunftsmodell: Patienten mit erhöhtem Risiko können durch Gentests und Familienanamnese identifiziert werden. Kardiale Bildgebung und Elektrophysiologie könnten bei dieser Untergruppe eine frühe Erkrankung erkennen. Durch die Kombination von Gentests und kardialer Phänotypisierung können Patienten mit hohem, mittlerem und niedrigem Risiko für personalisierte Empfehlungen stratifiziert werden. Zu den Patienten mit hohem Risiko gehören Patienten mit Synkopen in der Vorgeschichte (QTc ≥500 ms) und plötzlichem Herztod in der Familienanamnese.
Copyright bei Aerzteverlag medinfo AG
Witellikerstrasse 40
8032 Zürich
thomas.szucs@hin.ch
Der Autor hat keinen Interessenskonflikt in Zusammenhang mit diesem Artikel deklariert.
1. Musunuru K et al. Genetic Testing for Inherited Cardiovascular Diseases. A Scientific Statement From the American Heart Association. Circ Genom Precis Med. 2020;13:e000067. DOI: 10.1161/HCG.0000000000000067
2. Moog U., Riess O. Medizinische Genetik für die Praxis. Thieme Stuttgart 2014

info@herz+gefäss
- Vol. 12
- Ausgabe 1
- Februar 2022