
Cystische Fibrose (CF), ist die häufigste, autosomal-rezessiv vererbbare und nach wie vor lebenslimitierende Multisystemerkrankung in Europa und den USA. Dank fortschrittlicher Behandlungsmöglichkeiten und frühzeitiger Diagnostik ist die früher in jungen Jahren letal verlaufende Krankheit nun auch eine Erkrankung des Erwachsenenalters geworden. Deshalb ist es wichtig, dass Grundversorger mit dem heterogenen Krankheitsbild der CF und der zur Verfügung stehenden Diagnostik vertraut sind und erkennen, wann diese Diagnose in Betracht zu ziehen ist. Zudem gibt es Patienten, die erst im Erwachsenenalter zur Diagnose kommen, da sie eine CF-ähnliche, aber mildere oder eine Mono-Symptomatik zeigen, die jedoch auch durch Varianten im CFTR-Gen verursacht wird.
Cystic fibrosis (CF) is the most common, autosomal recessive, and still life-limiting multisystem disease in Europe and the USA. Thanks to advanced treatment options and early diagnosis, the disease, which used to be lethal at a young age, has now become an adult-onset disease. Therefore, it is important that primary care providers are familiar with the heterogeneous clinical picture of CF and the available diagnostics and recognize when this diagnosis should be considered. In addition, there are patients who do not come to diagnosis until adulthood because they present with CF-like but milder symptoms or mono-symptomatology, but this is also caused by variants in the CFTR gene.
Key Words: Cystische Fibrose, CFTR, CFTR-related disorders
Pathophysiologie
Bei der CF handelt es sich um eine Stoffwechselstörung, welche durch nicht oder nur teilweise funktionierende Chloridkanäle in der Membran sekretorischer Zellen verursacht wird. Durch die fehlende oder ungenügende Chloridsekretion, das dadurch reduzierte Austreten von Wasser aus den Zellen und die Zunahme der Natriumresorption kommt es zur Produktion von zähflüssigen, schwer abzutransportierenden Sekreten der endokrinen Drüsen. Folge davon sind Funktionsstörungen der Epithelien der betroffenen Organe, insbesondere der Atemwege, der Gallenwege, der Leber, der Pankreasgänge, des Dünndarms und der Samenleiter. Die Schweissdrüsen, welche ebenfalls Chloridkanäle besitzen, haben als Regulatoren der Körpertemperatur die Aufgabe, Chlorid in die Zellen aufzunehmen und nicht abzugeben, wodurch bei Fehlfunktion der Chloridkanäle der Schweiss salziger wird.
Diagnostik
Dank fortschrittlicher Behandlungsmöglichkeiten und frühzeitiger Diagnostik, liegt die durchschnittliche Lebenserwartung in der westlichen Welt bereits bei 50-55 Jahren, wobei CF früher bereits im Kindesalter letal verlief. Für Verlauf und Lebenserwartung ist eine frühe Diagnose und Behandlung aber entscheidend. Deshalb wurde in der Schweiz 2011 das Neugeborenen-Screening implementiert, das allerdings, wie der Name sagt, ein Screening- und nicht ein diagnostischer Test ist. Auf ein auffälliges Screening muss unmittelbar ein diagnostischer Test folgen. Der älteste Test und nach wie vor Goldstandard der CF-Diagnostik ist der sogenannte Schweisstest. Ergibt die Bestimmung des Chlorids im Schweiss mittels Pilocarpin-Iontophorese eine Chloridionen-Konzentration von ≤ 30 mmol/l ist eine CF unwahrscheinlich und bei 30-60 mmol/l ist der Befund nicht eindeutig (intermediär), bei ≥ 60 mmol/l jedoch ist die Diagnose einer CF sehr suggestiv. Bei pathologischem und intermediärem Schweisstest ist eine nachfolgende genetische Analyse obligatorisch, einerseits zur Bestätigung der Diagnose und andererseits um die adäquate mutationsspezifische Therapie anzuwenden. Im Bereich der Familienplanung kann Paaren, die beide gesunde CF-Träger (Heterozygote) sind, auf Wunsch eine Pränataldiagnostik (PnD) nach Chorionzottenbiopsie (10. bis 12. SSW) oder nach Amniozentese (14. bis 16. SSW) oder aber bei Inanspruchnahme einer reproduktionsmedizinischen Unterstützung eine Präimplantationsdiagnostik (PID) angeboten werden.

Genetik der CF
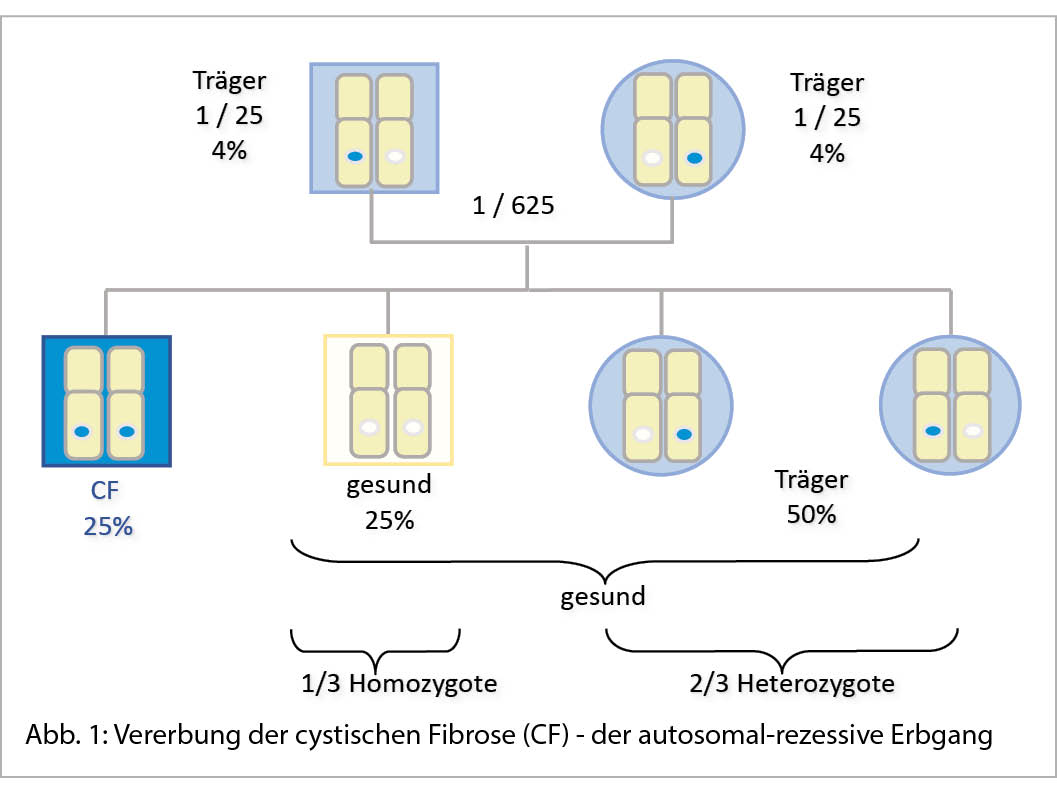
Vererbung und Häufigkeit (Abb.1)
Die CF wird autosomal-rezessiv vererbt, d.h. um zu erkranken muss eine Person sowohl im väterlichen wie auch im mütterlichen CF-Gen eine pathogene Veränderung (Variante, Mutation) tragen. Handelt es sich um zwei gleiche pathogene Varianten, so ist der Patient bezüglich dieser Variante homozygot. Sind in den beiden CF-Genen zwei verschiedene pathogene Varianten vorhanden, nennt man dies Compound-Heterozygotie. In der Schweiz ist ca. jede 25. Person heterozygot für eine CF verursachende Variante. Das bedeutet, dass in mindestens jeder 600. Partnerschaft beide Partner gesunde Träger einer CF-Variante sind, womit ein Risiko von 25% besteht, dass ein gemeinsames Kind des Paares von jedem Elternteil das mutierte CF-Gen erbt und an CF erkrankt. Mit einer Wahrscheinlichkeit von 75% werden die Kinder gesund sein und zwar entweder Träger wie ihre Eltern oder aber ganz gesund ohne CF-Mutation. Prospektiv haben also Eltern, die Träger sind ein Risiko von 50%, dass ihre Nachkommen heterozygot für eine CF-Variante sind, die dann wiederum weitervererbt werden kann. Möchte jedoch ein gesundes Geschwister eines CF-Patienten sein Trägerrisiko wissen, so beträgt dieses 67% (2/3).
Gen-, Genprodukt & Mutationsklassen (Tab. 1)
Im Jahr 1989 wurde das in mutiertem Zustand für CF verantwortliche Gen (CFTR-Gen) auf Chromosom 7 (7q31.2) entdeckt und charakterisiert (1). Mehr als 2100 verschiedene, über das ganze Gen verteilte Varianten wurden bisher in der kanadischen CF-Mutationsdatenbank aufgeführt (www.genet.sickkids.on.ca), wovon ca. 1200 CF verursachend sind und 466 funktionell untersucht wurden (www.cftr.org).
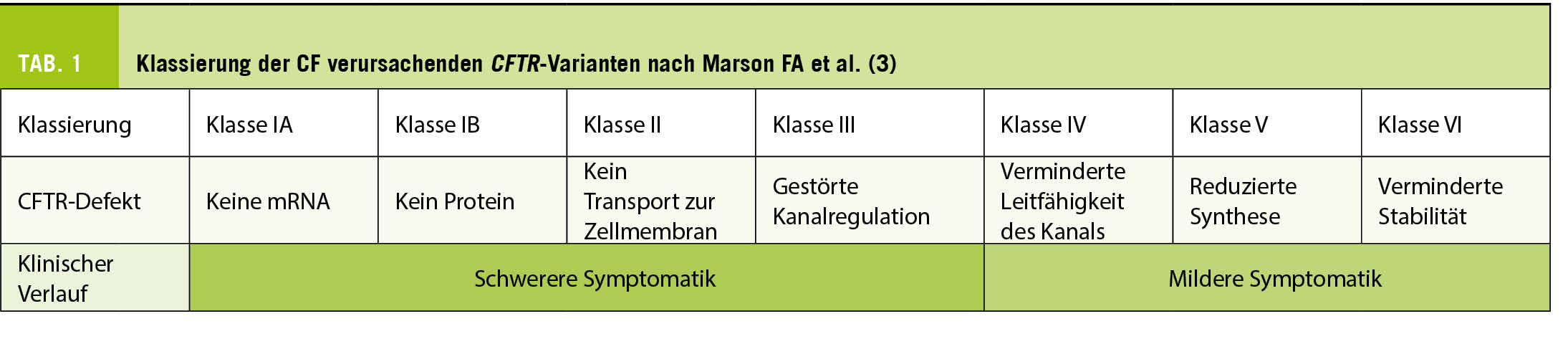
Nicht alle CF verursachenden Varianten beeinträchtigen die Funktion und/oder die Synthese des CFTR-Proteins gleichermassen, weshalb sie in verschiedene Klassen eingeteilt werden (2), die in Tabelle 1 dargestellt sind. Es ist aber durchaus möglich, dass eine pathogene Variante zu mehr als einer Klasse gehören kann. Die weltweit häufigste CF verursachende Variante F508del z.B. gehört zu Klasse II indem sie die korrekte Faltung des Proteins verhindert und damit dessen Degradation iniziiert. F508del-Proteine, die der Degradation entgehen und die Zellmembran erreichen zeigen
einen Regulierungs-Defekt des Chloridkanals (Klasse III) und reduzieren dessen Stabilität (Klasse IV).
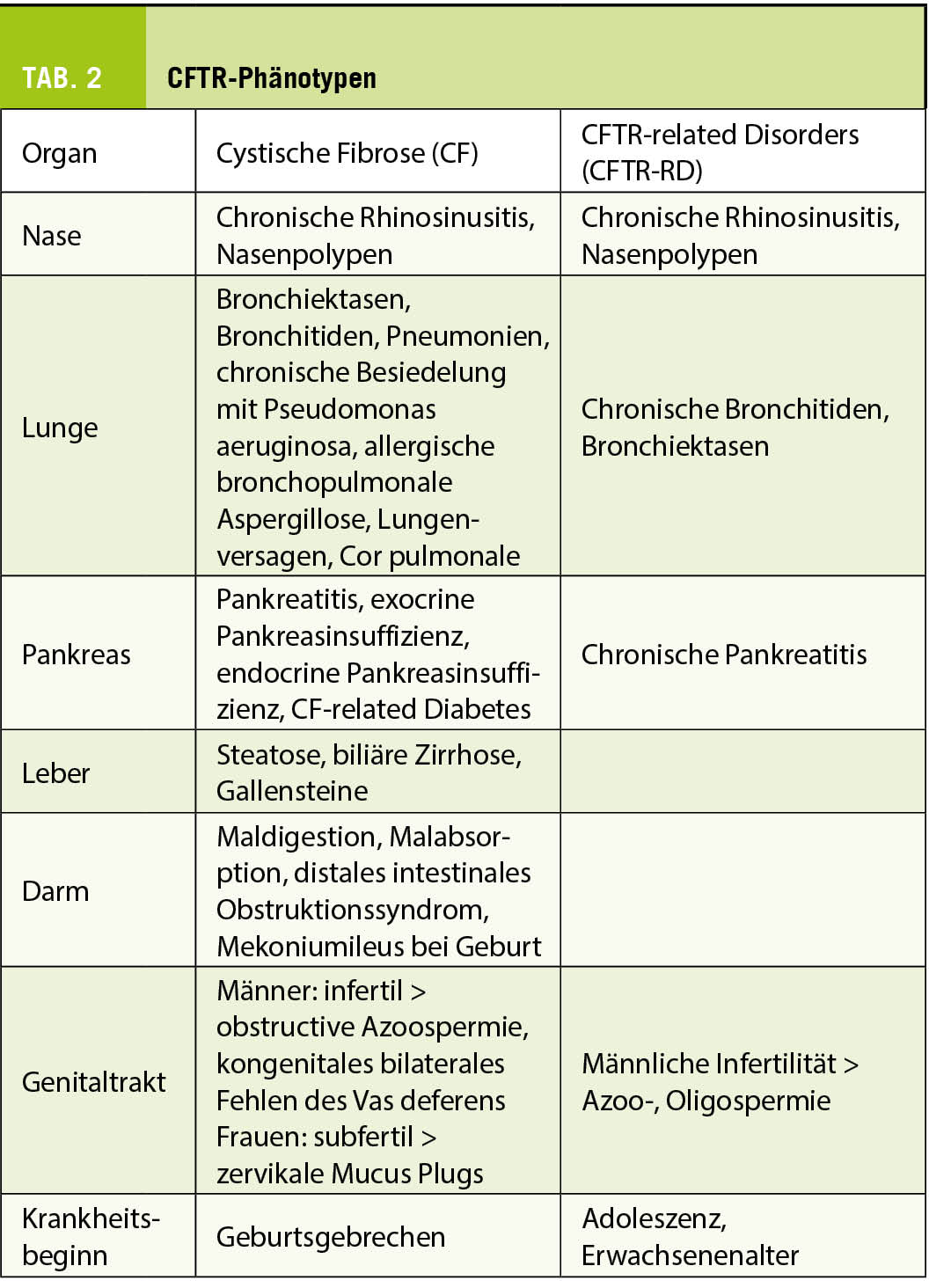
Heterogenität des Krankheitbildes (Phänotyp) (Tab. 2)
Die CF manifestiert sich mit einer enormen Variabilität und Komplexität, die zu einem grossen Teil davon abhängt, ob und wenn ja wieviel Restfunktion des Chloridkanals vorhanden ist. Bei der klassischen CF wird vor allem zwischen pankreasinsuffizienten (PI) und pankreassuffizienten Patienten (PS) unterschieden, wobei PS-Patienten einen milderen Verlauf zeigen als PI-Betroffene. Bei einer CF-ähnlichen, aber viel milderen und erst im Erwachsenenalter auftretenden Symptomatik sowie bei monosymptomatischen Erkrankungen wie chronische Pankreatitis, Bronchiektasen unklarer Aetiologie, chronische Bronchitis oder isolierte männliche Infertilität mit oder ohne CAVD (congenital absence of the vas deferens) und Nachweis von CFTR-Varianten, die nicht CF verursachen, wird die Diagnose eines CFTR-related disorder (CFTR-RD) gestellt. In Tabelle 2 sind die betroffenen Organe und die verschiedenen Symptome zusammengefasst.
Therapie
Eine kausale Therapie gibt es nach wie vor nicht für die CF, wohl aber mehrere Lebensqualität und Lebenserwartung steigernde Therapiemassnahmen. CF-Betroffene werden in der Regel alle drei Monate im CF-Zentrum gesehen, einerseits für das Monitoring des Krankheitsverlaufs und andererseits zur Überprüfung und allenfalls Modifikation der verschiedenen Therapien (3).
- Etablierte Therapien sind: Enzymersatz- und Ernährungstherapie zur Verhinderung einer Gedeihstörung, tägliche Inhalations- und Atemphysiotherapie zur Reinigung der Bronchien von zähem Sekret zur Erhaltung der Lungenfunktion sowie agressive Antibiotikatherapie bei Infekten, um möglichst lange eine chronische Besiedelung mit Pseudomonas aeruginosa zu verhindern.
- Neuer präzisionsmedizinischer Therapieansatz: CFTR-Modulatoren, die entsprechend der genetischen Konstellation (Genotyp spezifisch) eingesetzt werden können. Zur Anwendung kommen sog. Potentiatoren (z.B. Ivacaftor), welche die Chloridkanal-Aktivität und den Chloridionen-Transport verbessern, oder Korrektoren (z.B. Tezacaftor, Elexacaftor), die Faltung, Ausreifung und Transport des Proteins verbessern oder aber eine Kombination von beiden – je nach Auswirkung der pathogenen Varianten auf das CFTR-Protein. Potentiatoren werden bei Varianten der Klasse III und IV eingesetzt und eine Kombination von Potentiator und Korrektor bei Varianten der Klasse II. Für die Behandlung von Patienten mit einer oder zwei F508del-Varianten hat eine Triple-Therapie (Trikafta) mit Elexacaftor, Tezacaftor (Korrektoren) und Ivacaftor (Potentiator) den grössten Effekt gezeigt (4).

Copyright bei Aerzteverlag medinfo AG
Forchstrasse 452
CH-8702 Zollikon
sabina.gallati@hirslanden.ch
Die Autorin hat keinen Interessenskonflikt in
Zusammenhang mit diesem Artikel deklariert.
1. Riordan J, Rommens J, Kerem B, et al. Identification of the cystic fibrosis
gene: cloning and characterization of complementary DNA. Science 1989;245(4922):1066-1073.
2. Marson FA, Bertuzzo CS, Ribeiro JD. Classification of CFTR mutation classes.
Lancet Respir Med 2016;4(8):e37-e38.
3. Kerem E, Conway S, Elborn S et al. Standards of care for patients with cystic
fibrosis: a European consensus. J Cyst Fibros 2005;4(1):7-26.
4. Haq I, Almulhem M, Soars S et al. Precision medicine based on CFTR genotype for people with cystic fibrosis. Pharmgenomics Pers Med 2022;15:91-104.

der informierte @rzt
- Vol. 13
- Ausgabe 3
- März 2022