Die Hypertrophe (obstruktive) Kardiomyopathie (HCM/HOCM) ist eine genetisch bedingte Anlagestörung des Herzmuskels. Diagnostiziert wird die Krankheit mittels EKG, Echokardiographie, und kardialer Magnetresonanztomographie. Der Nachweis einer pathologischen Mutation lässt die Krankheit gegenüber anderen Krankheiten mit ähnlichem Phänotyp sicher abgrenzen und erlaubt allenfalls eine individualisierte Therapie.
Hypertrophic (obstructive) cardiomyopathy (HCM/HOCM) is a genetic disorder of the heart muscle. The disease is diagnosed by ECG, echocardiography, and cardiac magnetic resonance imaging. The detection of a pathological mutation allows the disease to be reliably distinguished from other diseases with a similar phenotype and, if necessary, permits individualized therapy.
Key Words: Hypertrophic (obstructive) cardiomyopathy, genetic risk, cardiogenetics, cardiomyopathy
Die HCM wird typischerweise verursacht durch Mutationen in Genen, welche für den kontraktilen Apparat kodieren. Dies führt zu einer Hypertrophie des linken Ventrikels mit ungewöhnlicher Anordnung der Myofibrillen. Abhängig von der Lokalität und der Ausprägung der Hypertrophie resultieren unterschiedliche Symptome aus dem Formenkreis der Herzinsuffizienz, der Thoraxschmerzen und der Arrhythmien. Die phänotypische Ausprägung beginnt meist in der Pubertät und ist interindividuell sehr unterschiedlich. Bei den meisten Patienten besteht ein benigner Verlauf (1). Allerdings können, wie unser Fallbeispiel zeigt, immer wieder auch sehr ausgeprägte Veränderungen beobachtet werden.
Fallbeispiel
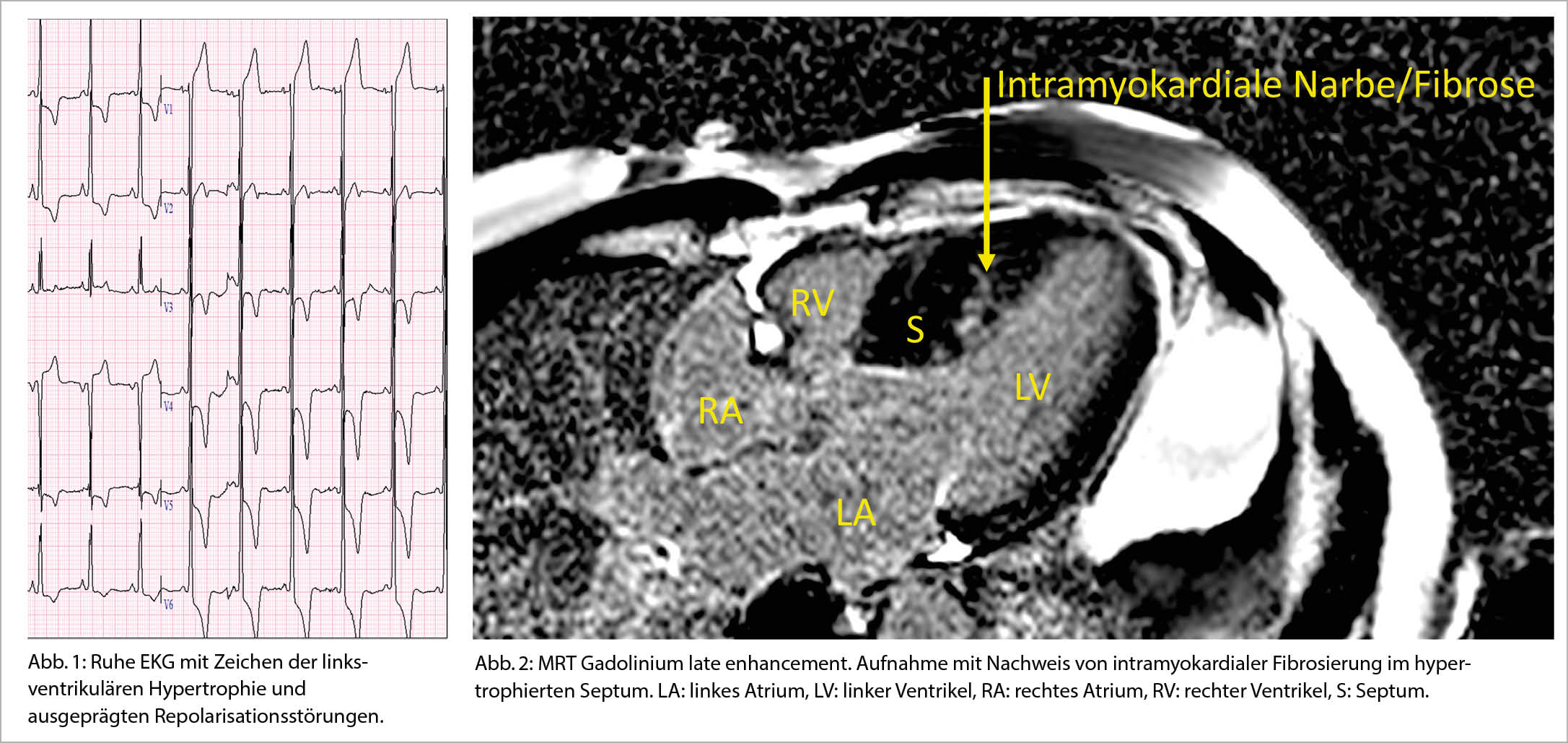
Der 49-jährige Patient war initial wegen einer Lumbago hospitalisiert. Anlässlich der klinischen Untersuchung liess sich ein auffälliges 3/6 Systolikum auskultieren, und es zeigte sich ein pathologisches Ruhe-EKG (Abb. 1). In der transthorakalen Farbdopplerechokardiographie zeigte sich eine schwere Hypertrophie des linken Ventrikels mit einer ausgeprägten Verdickung insbesondere des interventrikulären Septums bis 25 mm. Diese Hypertrophie führte zu einer systolischen Vorwärtsbewegung des vorderen Mitralsegels (systolic anterior movement, SAM) und als Folge davon zu einer Mitralklappeninsuffizienz, sowie zu einer Obstruktion im linksventrikulären Ausflusstrakt (LVOT). Die Diagnose einer HOCM wurde mit einer kardialen Magnetresonanz Tomographie (MRT) bestätigt, welche die schwere, septal betonte Hypertrophie bestätigte, und eine zusätzliche lokalisierte Hypertrophie inferoapikal objektivierte. Insbesondere in diesen hypertrophierten Arealen zeigte sich bei den «Late Gadolinium Enhancement» Sequenzen eine typische, diffuse intramyokardiale Fibrosierung (Abb. 2). Im 24-Stunden Holter-EKG zeigten sich keine relevanten Arrhythmien. Während ca. 5 Jahren bestand ein relativ stabiler Verlauf, der Patient beklagte jedoch eine zunehmende Anstrengungsdyspnoe und schliesslich auch relativ typische pectanginöse Beschwerden, welche sich durch diverse Medikamente (Betablocker, Ca-Antagonisten, Ranolazin) nicht adäquat behandeln liessen. Eine koronare Herzkrankheit wurde angiographisch ausgeschlossen. Echokardiographisch wurde eine weitere Progredienz der Befunde registriert. Es entwickelten sich schliesslich eine schwere Obstruktion im LVOT mit maximalen Druckgradienten unter Valsalva bis >200 mmHg, sowie durch den SAM auch eine schwere Mitralklappeninsuffizienz. Zur Verminderung des Druckes im LVOT wurde eine transkoronare Ablation der Septumhypertrophie (TASH) durchgeführt, welche jedoch nur kurz zu einer leichten Abnahme des Druckgradienten im LVOT, und zu keiner Besserung der Klinik führte. Aus diesem Grund erfolgte schliesslich eine chirurgische Myektomie und ein Mitralklappenersatz. Da es postoperativ zu einem kompletten AV-Block kam musste auch noch ein Schrittmacher implantiert werden. Seit der abgeschlossenen Rehabilitation zeigt der Patient nun jedoch einen erfreulichen Verlauf und auch die pectanginösen Beschwerden sind vollständig verschwunden.
Genetische Abklärung
Der Patient hat drei Brüder und zwei Schwestern, die, soweit bekannt, keine kardialen Probleme haben. Bei zwei Brüdern fiel die kardiologische Untersuchung unauffällig aus. Der Vater des Patienten verstarb 74-jährig wahrscheinlich an einer terminalen Herzinsuffizienz, die Mutter ist bisher gesund. Bei einem der beiden Söhne besteht im Alter von 19 Jahren ebenfalls bereits eine schwere linksventrikuläre Hypertrophie. Beim anderen Sohn wurde im Alter von 24 Jahren eine leichte linksventrikuläre Hypertrophie festgestellt. Mit dem Patienten und dem sicher betroffenen Sohn wurden die Möglichkeiten und Grenzen einer molekulargenetischen Analyse eingehend besprochen. Mit deren Einverständnis, sowie nach Einholen einer Kostengutsprache bei der Krankenkasse, wurde eine Genanalyse veranlasst. Mittels Hochdurchsatz-Sequenzierung wurden simultan 78 Kardiomyopathie-Gene sequenziert und analysiert. Es zeigten sich sowohl beim Indexpatienten, wie auch bei seinem Sohn eine Sequenzvariante im MYH7-Gen (c.1324C>7 p.Arg442Cys).
Besprechung
Die identifizierte Sequenzvariante wurde bereits mehrmals bei Patienten mit HCM beschrieben, weshalb von einer Klasse 4 Variante (likely pathogenic) gemäss «American College for Medical Genetics and Genomics» ausgegangen werden kann. Allerdings gibt es für diese MYH7-Genvariante keine Genotyp - Phänotyp Korrelation, welche den individuellen Verlauf der HCM voraussagen, und eine noch optimalere kardiologische Betreuung des Patienten erlauben würde. Der Hauptbenefit der Untersuchung besteht somit darin, dass alle Familienangehörigen mit erhöhtem Risiko für eine HCM auf Wunsch die Möglichkeit eines prädiktiven genetischen Tests haben (Kaskadenscreening). Nicht-Mutationsträger können von regelmässigen kardiologischen Untersuchungen befreit werden, währendem sich Mutations-Träger weiterhin regelmässig kardiologisch (EKG und Echokardiographie) untersuchen lassen sollten. Dies insbesondere auch vor dem Hintergrund, dass mit Mavacamten möglicherweise demnächst ein Medikament zur Verfügung stehen wird, mit dem auch der Krankheitsverlauf günstig beeinflusst werden kann (2).
Das MYH7-Gen, welches für die Beta-myosin heavy chain kodiert, ist neben dem MYBPC3-Gen, das für Myosin binding protein C kodiert, eines der beiden Hauptgene der erblich bedingten HCM (3). Diese beiden Gene sind für ca. 40% der familiären HCM verantwortlich. Die Beta-myosin heavy chain Proteine werden in den Myozyten des Herzens und den langsamen Fasern des Skelettmuskels exprimiert. Mehrere hundert «pathogene Varianten» (PV) und «wahrscheinlich pathogene Varianten» (LPV, likely pathogenic variants) wurden in Patienten mit HCM identifiziert. Bei der in unserem Fall beobachteten Mutation handelt es sich um eine «missense mutation». Es gilt zu beachten, dass auch pathogene Varianten in ganz anderen Genen zu einem sehr ähnlichen klinischen Bild führen können, man spricht von sogenannten Phänokopien. Bei der HCM ist dabei insbesondere an den Morbus Fabry, andere lysosomale Speicherkrankheit, oder an ein Noonan Syndrom (Gendefekt im PTPN-11 Gen) zu denken. Insofern liefert die genetische Abklärung wichtige Zusatzinformationen, welche auch relevante Implikationen für die Therapie haben. Andererseits schliesst ein negativer Gentest eine HCM nicht aus.
Copyright bei Aerzteverlag medinfo AG
Facharzt für Kardiologie FMH
Schänzlistrasse 33
3013 Bern
peter.burger@hin.ch
Der Autor hat keinen Interessenskonflikt in Zusammenhang mit diesem Artikel deklariert.
1. UpToDate; Hypertrophic cardiomyopathy
2. Olivatto et al. Lancet 2020; 396: 759–69
3. Marian AJ. Circulation Research. 2021;128:1533–1553.
