
Molekularpathologische Untersuchungen des Tumorzellgenoms können Befunde liefern, die auf eine genetische Tumorprädisposition hindeuten. Dieser Artikel beschreibt molekulare Testergebnisse, bei denen genetische Beratung und Keimbahntestung mit dem Patienten diskutiert werden sollten.
Tumor genomic testing may deliver results that are indicative of a genetic tumor predisposition. This article describes molecular test results that should lead to a discussion with the patient on genetic counselling and germline testing.
Key Words: Erbliche Tumorprädisposition, Molekularpathologie, Genetische Beratung, Keimbahntestung
10–20% aller Krebserkrankungen entstehen im Rahmen einer erblichen Tumorprädisposition. Ursächlich beteiligte Gene, Vererbungsmodus und klinischer Phänotyp der häufigen Tumorprädispositionssyndrome sind inzwischen bekannt und in der
Online Mendelian Inheritance in Man (OMIM) Datenbank umfassend dokumentiert.
Aktuelle molekularpathologische Untersuchungen dienen dem Nachweis von Veränderungen im Tumorzellgenom, die als diagnostische, prognostische oder therapeutisch-prädiktive Biomarker bei onkologischen Erkrankungen klinische Anwendung finden. Veränderungen im Tumorzellgenom können direkt oder indirekt mit verschiedenen Methoden auf unterschiedlichen biochemischen Ebenen (DNA, RNA, Protein) detektiert werden. Die hochparallele Sequenzierung (Next Generation Sequencing, NGS) hat sich hierfür als Schlüsselverfahren auch für die molekularpathologische Routinediagnostik etabliert.
Genetische Beratungen werden bei Verdacht auf eine erbliche Erkrankung vor einer Keimbahntestung und nach Vorliegen der Testergebnisse durchgeführt. Aktuelle Schweizer Richtlinien für Patienten mit Mamma-, Ovarial-, Pankreas- oder Prostatakarzinom empfehlen genetische Beratung und Keimbahntestung in Hinblick auf eine erbliche Tumordisposition vor allem auf der Grundlage einer positiven persönlichen oder Familienanamnese (1). Das genetische Beratungsgespräch soll einen Patienten in die Lage versetzen, eine informierte Entscheidung für oder gegen eine Keimbahntestung in Hinblick auf eine erbliche Erkrankung zu treffen. Weiterhin dient es dazu, die Einwilligung der betroffenen Person zu einer Keimbahntestung einzuholen. Bei einem positiven Testergebnis informiert die genetische Beratung über weitere Tumorrisiken, mögliche Vorsorgeuntersuchungen, das Vererbungsrisiko und die gezielte genetische Testung von Familienangehörigen.
Dieser Beitrag beschreibt typische molekularpathologische Befundsituationen, die auf eine mögliche erbliche Tumordisposition hindeuten und bei denen genetische Beratung und Keimbahntestung in Hinblick auf ein genetisches Tumordispositionssyndrom indiziert sind.

Pathogene Sequenzvariante in einem Tumorprädispositionsgen
Die meisten zur NGS-basierten Tumortestung eingesetzten Genpanel umfassen Gene, die gemäss OMIM Datenbank mit einer erblichen Tumorprädisposition assoziiert sind. Für die Klassifikation von (somatischen und Keimbahn-) Varianten werden fünf Kategorien verwendet (benigne, wahrscheinlich benigne, Variante unklarer Signifikanz, wahrscheinlich pathogen, pathogen) (2). Bei rund 20% der bei einer Tumortestung in einem Tumorprädispositionsgen nachgewiesenen pathogenen Sequenzvarianten handelt es sich um eine Keimbahnvariante (3). Ohne vergleichende Untersuchung von Nichttumorgewebe (oder Blutzellen) des gleichen Individuums kann nicht sicher unterschieden werden, ob es sich bei einem positiven Testergebnis um eine bei der Tumorentstehung erworbene (somatische) Variante oder eine (konstitutionelle) Keimbahnvariante handelt. Hinweise auf eine Keimbahnvariante ergeben sich aus der Allelfrequenz der Sequenzvariante, dem Vorliegen einer «Gründermutation» und dem Nachweis der gleichen Sequenzvariante in mehreren Tumoren eines Patienten.
Bei pathogenen Varianten mit einer Allelfrequenz um 50% (30-70%) wird ein Ursprung in der Keimbahn angenommen. Je höher die Allelfrequenz in der Tumortestung, desto höher ist die Wahrscheinlichkeit einer Keimbahnvariante. Dabei ist zu beachten, dass die Allelfrequenz durch den Tumorzellgehalt des Probenmaterials, intratumorale Heterogenität (Existenz von verschiedenen Klonen innerhalb eines Tumors) und gleichzeitig vorliegende nummerische Genveränderungen beeinflusst wird.
Als Gründermutation wird eine Keimbahnvariante bezeichnet, die mit grosser Häufigkeit in einer definierten Population beobachtet wird. Für mehrere Tumorprädispositionsgene (u.a. BRCA1, BRCA2, TP53) sind pathogene Keimbahnvarianten bekannt, die besonders häufig in bestimmten geographischen Regionen oder in bestimmten Bevölkerungsgruppen vorkommen.
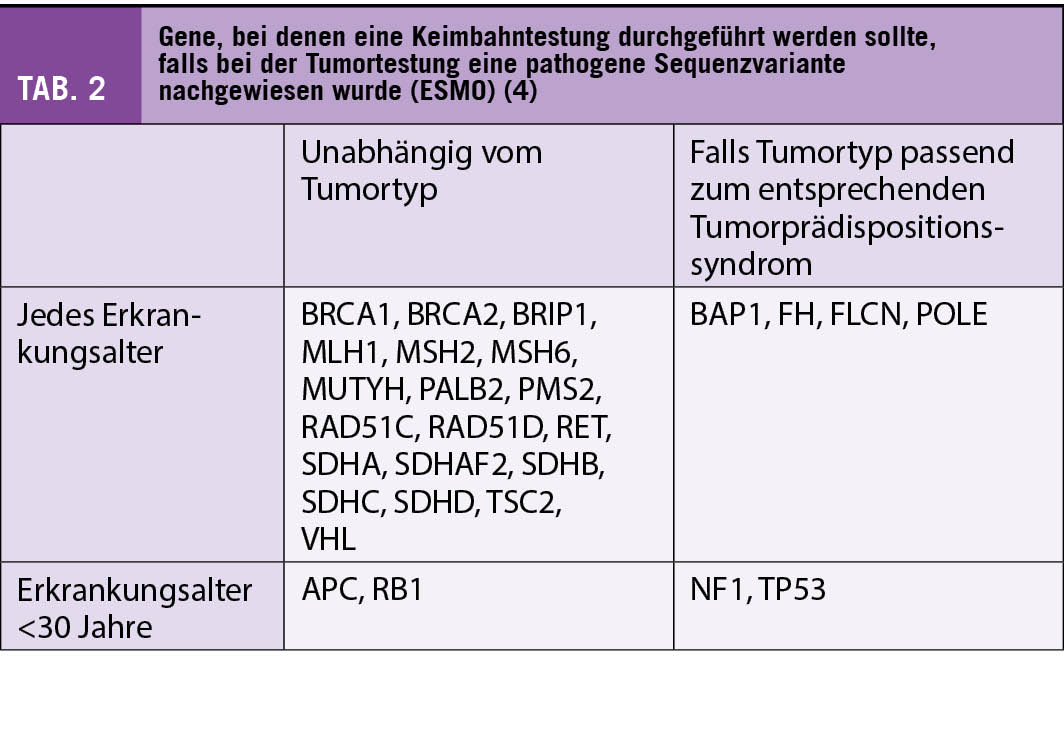
Eine genetische Beratung sollte durchgeführt werden, wenn im Tumorzellgenom eine pathogene Variante in einem Tumorprädispositionsgen vorliegt (1). Dies gilt besonders für Tumorprädispositionsgene, bei denen eine pathogene Keimbahnvariante mit einer hohen Tumorpenetranz assoziiert ist und/oder klinische Optionen zur Verminderung des Tumorerkrankungsrisikos bestehen (Früherkennung, chirurgische Massnahmen, etc.). Die Precision Medicine Working Group der Europäischen Gesellschaft für medizinische Onkologie (ESMO) hat eine Liste von 27 Genen veröffentlicht, bei denen der Nachweis einer pathogenen Sequenzvariante in der Tumortestung zu einer Keimbahntestung führen sollte (Tab. 2) (4).
Defekte DNA-Mismatch-Reparatur
Bei Ausfall der DNA-Mismatch-Reparatur (MMR) werden Basenfehlpaarungen im Genom nicht mehr korrigiert. Dies führt zu einer Anhäufung von somatischen Sequenzvarianten und zu Längenveränderungen der Mikrosatelliten (Mikrosatelliten-Instabilität, MSI). MMR-Defekte werden durch einen Funktions- oder Expressionsverlust der MMR-Proteine MSH2, MSH6, MLH1 oder PMS2 verursacht. Ein Expressionsverlust von MMR-Proteinen (dMMR) kann mittels Immunhistochemie nachgewiesen werden, Längenveränderungen der Mikrosatelliten durch molekulare Verfahren. MMR-Expressionsverlust und hochgradige MSI in Tumorzellen können auf ein Lynch-Syndrom hindeuten, ein erbliches Tumorprädispositionssyndrom, welches durch eine Keimbahnveränderung in einem MMR-Gen (meistens MLH1 und MSH2, selten MSH6 und PMS2) oder eine Deletion des EPCAM Gens verursacht wird. In Abhängigkeit vom Tumortyp kann bei bis zu 38% der Tumore mit hochgradiger MSI mit dem Vorliegen eines Lynch-Syndroms gerechnet werden (5). In der genannten Studie lag bei rund der Hälfte der Patienten mit gesichertem Lynch-Syndrom ein Tumortyp vor, der nicht zum typischen Tumorspektrum eines Lynch-Syndroms gehört und bei denen aufgrund der üblichen Kriterien keine Keimbahntestung empfohlen worden wäre.
Homologe Rekombinationsdefizienz
Als homologe Rekombinationsdefizienz (HRD) wird der funktionelle Ausfall der homologen Rekombinationsreparatur (HRR), einem zellulären Reparaturmechanismus zur Beseitigung von erworbenen DNA-Schäden, bezeichnet (6). Die HRD führt zu charakteristischen Veränderungen im Tumorzellgenom (genomische Narbe), die mittels bioinformatischer Algorithmen klassifiziert und quantifiziert werden können (u.a. Genomic Instability Score, HRDetect). Von klinisch-therapeutischer Bedeutung ist die Assoziation zwischen HRD und dem Ansprechen auf Poly(ADP-Ribose)-Polymerase (PARP)-Inhibitoren und Platinum-basierte Chemotherapien bei verschiedenen Tumortypen (Ovarial-, Mamma, Pankreas- und Prostatakarzinom). Die HRD wird häufig durch eine BRCA1 oder BRCA2 Inaktivierung ausgelöst, kann aber auch durch pathogene Varianten in anderen HRR-assoziierten Genen verursacht werden. Keimbahnvarianten dieser HRR-assoziierten Gene (v.a. BRCA1, BRCA2) sind mit einer erblichen Tumordisposition assoziiert. Somit kann der Nachweis einer HRD auf eine genetische Tumordisposition hindeuten.
Hypermutierte Tumore
Hypermutierte Tumore zeigen eine hohe Tumormutationslast (TML), d.h. eine grosse Anzahl von erworbenen Sequenzvarianten im Tumorzellgenom. Bei mehreren Tumortypen (u.a. Endometriumkarzinom, kolorektales Karzinom, Melanom) ist ein hypermutierter Status mit einem Ansprechen auf Immuncheckpoint-Inhibitoren assoziiert. Neben einem Ausfall der DNA-Mismatch-Reparatur (siehe oben) führt vor allem eine somatische Inaktivierung der POLD1 und POLE Gene zu einer hohen TML. Im Gegensatz dazu sind pathogene POLD1 und POLE Keimbahnvarianten sehr selten und gehen mit einem erhöhten Erkrankungsrisiko für kolorektale Karzinome, Endometriumkarzinome und weitere Tumortypen einher (7). Auch wenn POLD1 und POLE Keimbahnvarianten seltene Ursachen einer erblichen Tumordisposition darstellen (0,1-0,4%), sollte bei einem Patienten mit hypermutiertem Tumor bei erhaltender DNA-Mismatch-Reparatur eine POLD1/POLE Keimbahntestung in Erwägung gezogen werden (7).
Sekundäre (inzidentelle) Befunde
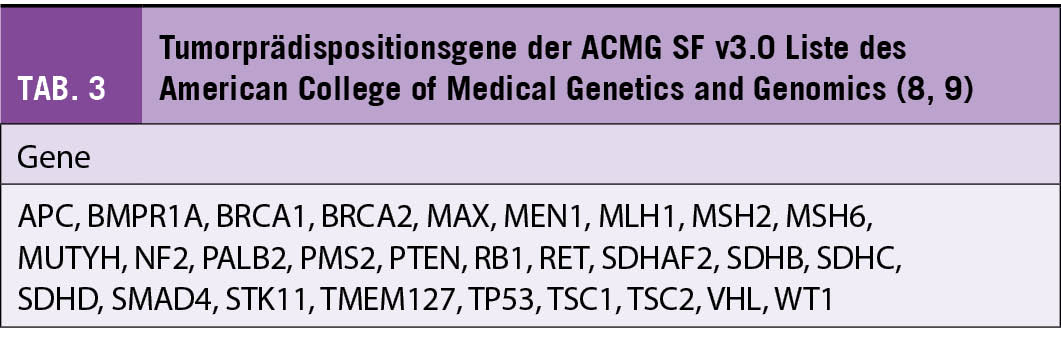
Als sekundäre (inzidentelle) Befunde gelten Ergebnisse, die ausserhalb der ursprünglichen Absicht bzw. Fragestellung einer Testung entdeckt werden (3). Verschiedene Fachgesellschaften haben Empfehlungen zum Umgang mit sekundären Keimbahnbefunden bei klinischen Exom- und Genomsequenzierungen veröffentlicht. Eine aktuelle Empfehlung des American College of Medical Genetics and Genomics (ACMG) enthält eine Liste von 73 Genen (ACMG SF v3.0), bei denen pathogene und wahrscheinlich pathogene Varianten auch dann berichtet werden sollten, wenn die Keimbahntestung mit einer anderen Fragestellung durchgeführt wurde (8, 9). Die ACMG Liste umfasst auch 28 Gene, die mit einer Tumorprädisposition assoziiert sind (Tab. 3). Bei Exom- und Genomsequenzierungen im Rahmen der molekularpathologischen Tumordiagnostik sind vor allem sekundäre Befunde von Bedeutung, die für Phänotypen ursächlich sind, die in keinem direkten Zusammenhang mit der onkologischen Erkrankung stehen. So nennt die ACMG Liste unter anderem Gene, die mit kardiovaskulären Erkrankungen, Stoffwechselstörungen und neurologischen Erkrankungen assoziiert sind.
Ausblick
Der Bedarf an genetischen Beratungen bei onkologischen Erkrankungen wird weiter zunehmen. Die nur begrenzt verfügbaren Beratungskapazitäten werden als Herausforderung bleiben.
In naher Zukunft wird das revidierte Gesetz über genetische Untersuchungen beim Menschen (GUMG) in Kraft gesetzt. Es enthält unter anderem neue Regelungen bezüglich Patientenaufklärung und Einwilligung für Tumortestungen, bei denen Befunde zu vererbbaren Eigenschaften (Überschussinformation) entstehen können.
Die Umsetzung der neuen regulatorischen Vorgaben erfordert organisatorische Anpassungen an der Schnittstelle zwischen Onkologie und Pathologie und höhere fachliche Anforderungen an Ärzte, die molekularpathologische Untersuchungen beauftragen und Untersuchungsergebnisse interpretieren.
Copyright bei Aerzteverlag medinfo AG
Institut für Medizinische Genetik der Universität Zürich
Schlieren
azzarello-burri@medgen.uzh.ch
Institut für Pathologie
Kantonsspital St. Gallen
9007 St. Gallen
wolfram.jochum@kssg.ch
Die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Beitrag deklariert.
1. Stoll S, Unger S, Azzarello-Burri S, Chappuis P, Graffeo R, Pichert G, et al. Update Swiss guideline for counselling and testing for predisposition to breast, ovarian, pancreatic and prostate cancer. Swiss Med Wkly. 2021;151:w30038.
2. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24.
3. Cushman-Vokoun A, Lauring J, Pfeifer J, Olson DR, Berry A, Thorson J, et al.
Laboratory and Clinical Implications of Incidental and Secondary Germline
Findings During Tumor Testing. Arch Pathol Lab Med. 2022;146(1):70-7.
4. Mandelker D, Donoghue M, Talukdar S, Bandlamudi C, Srinivasan P, Vivek M, et al. Germline-focussed analysis of tumour-only sequencing: recommendations from the ESMO Precision Medicine Working Group. Ann Oncol. 2019;30(8):1221-31.
5. Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, et al.
Microsatellite Instability Is Associated With the Presence of Lynch Syndrome
Pan-Cancer. J Clin Oncol. 2019;37(4):286-95.
6. Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol. 2020;31(12):1606-22.
7. Mur P, Garcia-Mulero S, Del Valle J, Magraner-Pardo L, Vidal A, Pineda M, et al. Role of POLE and POLD1 in familial cancer. Genet Med. 2020;22(12):2089-100.
8. Miller DT, Lee K, Chung WK, Gordon AS, Herman GE, Klein TE, et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome
sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(8):1381-90.
9. Miller DT, Lee K, Gordon AS, Amendola LM, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome
sequencing, 2021 update: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23(8):1391-8.


info@onco-suisse
- Vol. 12
- Ausgabe 6
- September 2022