Herzrhythmusstörungen, Herzinsuffizienz oder Diabetes können verschiedene Ursachen haben, wobei vielfach der Lebensstil bedeutend ist. Aber auch die genetische Prädisposition kann eine Rolle spielen. In diesem Beitrag wird eine Patientin vorgestellt, bei der als Grundkrankheit eine myotone Dystrophie Typ II (DM2) klinisch vermutet und molekulargenetisch nachgewiesen wurde. Es handelt sich somit um eine monogen verursachte Krankheit, die zu pathologischen Störungen in verschiedenen Organen, also einer Multisystemerkrankung, führt.
Cardiac arrhythmias, heart failure or diabetes can have various causes, whereby lifestyle is often significant. However, genetic predisposition can also play a role. This article presents a patient in whom myotonic dystrophy type II (DM2) was clinically suspected and molecularly proven as the underlying disease. Thus, it is a monogenic disease leading to pathological disturbances in different organs, i.e. a multisystem disease.
Key Words: myotonic dystrophy type II, genetic predisposition, monogenic disease
Fallbeispiel
Ausgangslage
Eine 41-jährige Ratsuchende wurde von ihrer Neurologin mit der Verdachtsdiagnose «proximal betonte Myopathie; DD: Myotone Dystrophie Typ II (DM2)» in die genetische Beratung überwiesen. Anfänglich fiel einzig eine leichte Hörstörung auf. Sie beklagte sich, dass sie seit etwa 10 Jahren zunehmend an Muskelschwäche der Oberarme und der Beine leide, so dass sie heute nur mit Schwierigkeiten auf einen Stuhl steigen könne, respektive bereits beim Aufstehen von einem solchen etwas Schwierigkeiten habe. Auch das Abstellen, respektive das Loslassen, der Einkaufstasche mache ihr Mühe. Zudem würden gelegentlich heftige Muskelschmerzen und alle 2 bis 3 Tage ein beängstigendes Herzrasen auftreten. Vor einigen Jahren wurde bei ihr zudem ein Diabetes mellitus diagnostiziert und kürzlich nach einem Skiunfall zufällig eine Verminderung des von Willebrand-Faktors (VWD Typ1) festgestellt. Ferner neige sie zu Verstopfung. Sie war nie schwanger. Die Antikonzeption erfolgte mit der Mirena-Hormonspirale. Für ihre Gesundheit ungünstige Umwelteinflüsse liessen sich nicht eruieren. Sie trinkt praktisch keinen Alkohol und raucht nur gelegentlich eine Zigarette.
Die gesundheitsbezogene Familienanamnese haben sie und ihr Vater sorgfältig in das Stammbaumschema eingetragen, das wir ihr vorgängig zugesandt hatten (1). Die Mutter, ein Einzelkind, verunfallte tödlich wegen eines Sturzes vom Fahrrad im Alter von 26 Jahren. Die beiden Stiefbrüder väterlicherseits sind gesund. Der Grossvater mütterlicherseits verstarb 41-jährig. Angeblich hatte auch er Diabetes und Herzprobleme. Die Grossmutter wurde 87 Jahre alt. In der Familie des Vaters lassen sich keine besonderen Gesundheitsprobleme feststellen.
Entscheid über genetische Analyse
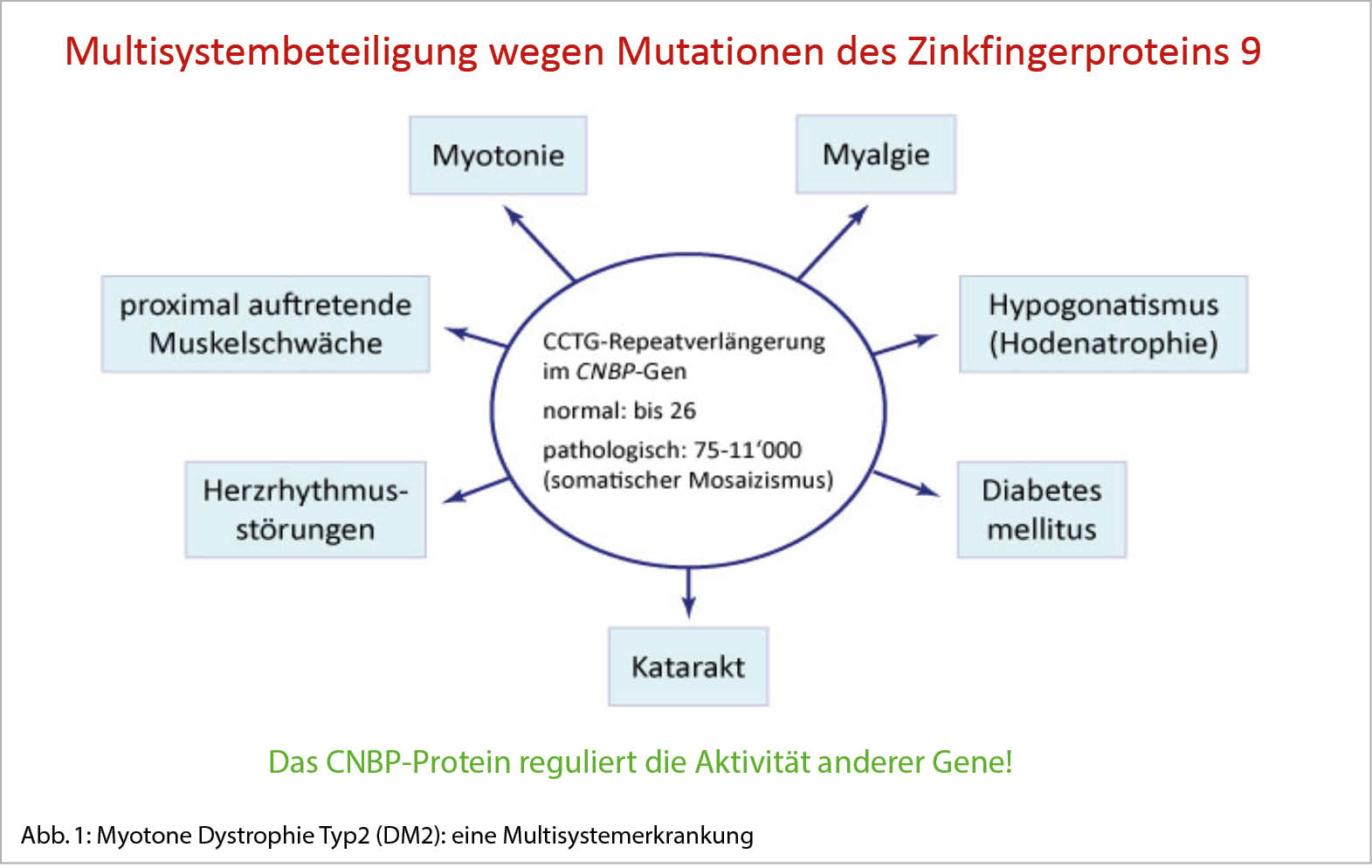
Die anlässlich der Beratung erhobenen Befunde sprechen für das Vorliegen der myotonen Dystrophie Typ 2 (DM2), einer monogenen Erbkrankheit, die sich auf eine Mutation des CNBP-Gens zurückführen lässt (Abb. 1). Die Myotonie und Dysfunktion der Muskeln sind die Kardinalsymptome. Auch Herzrhythmusstörungen/Herzleitungsstörungen (im Routine EKG als atrioventrikuläre Leitungsstörungen nachweisbar) treten als typische Komplikation auf. Diese können eine dilatierte Kardiomyopathie als Folge haben. Der Diabetes mellitus der Ratsuchenden lässt sich auf eine Insulininsensitivität zurückführen. Die Verschlechterung des Hörvermögens erinnert an eine vorzeitig eingetretene Presbyakusis (Altersschwerhörigkeit).
Dank der Kostengutsprache der Krankenkasse und mit dem schriftlichen Einverständnis der Ratsuchenden wurde die molekulargenetische Analyse des CNBP-Gens veranlasst.
Befund
Es wurde eine pathogene Expansion von CCTG-Repeats im ersten Intron eines der beiden CNBP-Gen festgestellt. Dabei handelt es sich um die einzige bekannte Ursache der DM2 (früheres Synonym PROMM («Proximal Myotonic Myopathy»)).
Differentialdiagnostisch muss das Vorliegen der häufigeren myotonen Dystrophie Typ 1 (DM1), gerade deren milde Form, erwogen werden, die sich auf Mutationen des DMPK-Gens zurückführen lässt. Deren kongenitale und kindliche Formen (herkömmliche Bezeichnung: Curshmann-Steinert-Syndrom) manifestiert sich im Gegensatz zur DM2 schon früh im Leben durch Entwicklungsstörungen, Atem- und Trinkprobleme, einer Verkürzung der Achillessehne sowie einer Intelligenzminderung. Im Falle eines negativen Resultats des CNBP-Gentests wäre ein Genpaneltest für neuromuskuläre Krankheiten indiziert gewesen, der mindestens folgende Gene umfassen sollte: DES, GNE, LDB3, MYOT, VCP (2).
Die VWD Typ 1 wurde molekulargenetisch nicht weiter abgeklärt, da sie keine gesundheitlichen Probleme verursacht und in der Regel auf Missense-Mutationen des Gens für den Von-Willebrand-Faktor (VWF) zurückzuführen ist. Sie wird autosomal-dominant vererbt.
Zur Genetik der DM2: Das CNBP-Gen kodiert ein sogenanntes Zinkfingerprotein. Dabei handelt es sich um eine Klasse von meistens regulatorisch wirkenden Proteinen, die sich an DNA oder RNA binden. Bei der DM2 liegt eine Wiederholung (Repetition) der (TG)n(TCTG)n(CCTG)n-Sequenz im CNBP-Gen vor, die eine pathogene Auswirkung hat. Dies führt zu einem Funktionsgewinn der RNA, die ihrerseits diejenige des Chloridkanals, des kardialen Troponins T oder des Insulinrezeptors beeinflusst (3). Wegen Schwierigkeiten der DNA-Verdoppelung solcher Sequenzen vor einer Zellteilung sind Veränderungen der «Repeat»-Zahl, d.h. somatische Mutationen, häufig. Missense-Mutationen wurden bei der DM2 nicht beobachtet. Die DM2 wird typischerweise autosomal-dominant vererbt. Ob möglicherweise die Mutter der Ratsuchenden eine Expansion hatte, bleibt ungeklärt. Ein mit den Methoden zur Analytik der DM2 vertrautes Labor hätte mit hoher Zuverlässigkeit die molekulargenetisch gesicherte Diagnose stellen können.
Bedeutung der genetischen Diagnose
Die umfassende medizinische Betreuung von Personen mit einer DM2-Veranlagung ist anspruchsvoll und komplex. Detaillierte Angaben über die Behandlung von Manifestationen der DM2 können in den neuen internationalen Pflegeempfehlungen entnommen werden (2). Fachärztinnen/-ärzte verschiedener Disziplinen sind bei der Betreuung von DM2-Patienten gefordert. Diese beinhaltet das Myalgie-Management (die Behandlung der Schmerz-Attacken) sowie jährlich ein Echokardiogramm und eine 24-stündige Holter-Überwachung zur Erfassung der Herzleitungsstörungen. Ein Herz-MRI kann zur frühzeitigen Erfassung einer Kardiomyopathie eingesetzt werden. Zudem ist die Kontrolle des Diabetes durch die jährliche Bestimmung der Glucosekonzentration im Serum sowie die Erfassung weiterer endokrinologischer Störungen (z.B. Funktionsstörungen der Schilddrüsen) indiziert. Auch eine jährliche augenärztliche Untersuchung ist bei Personen mit einer Abnahme des Sehvermögens empfehlenswert. Hörhilfen können bei sensoneurinalen Gehöreinbussen nützlich sein.
Copyright bei Aerzteverlag medinfo AG
Schönbeinstrasse 40
4031 Basel
hansjakob.mueller@unibas.ch
Der Autor hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. unispital-basel.ch/medizinische-genetik (Reiter: Formulare Medizinische Genetik)
2. Schoser B et al. Consensus-based recommendations for adults with myotonic dystrophy type 2. Neurol Clin Pract 2019; 9: 343-353
3. Thornton CA et al. Myotonic dystrophy: approach to therapy. Curr Opin Genet Dev 2017; 44: 135-140
4. Antonarakis SE. Krankheitsgenen auf der Spur! Eine kurze Geschichte der Methoden zu ihrer Identifikation. In: Müller Hansjakob, Hadorn Hans-Beat (Hg): Humangenetik und Anthropologie. Ein Zeitdokument. Schwabe-Verlag Basel 2022. pp 9-21

info@herz+gefäss
- Vol. 12
- Ausgabe 5
- Oktober 2022