
Im Dezember 2020 liess Swissmedic die «Orphan Drug» Trikafta® zu. Dabei handelt es sich um eine hochwirksame Kombination von Modulatoren des CFTR-Proteins (Cystic fibrosis transmembrane conductance regulator) zur Behandlung von Mukoviszidose bei Patienten mit mindestens einer F508del-Mutation im CFTR-Gen, der häufigsten Mutation bei Mukoviszidose. Die Wirkstoffe sind Elexacaftor, Tezacaftor und Ivacaftor. Derzeit kommen mehr als 85% der Patienten mit Mukoviszidose für eine Behandlung mit CFTR-Modulatoren in Frage, die die Lungenfunktion und die Lebensqualität verbessern und die Exazerbationen der Atemwege verringern.
In December 2020, Swissmedic approved the drug Trikafta®, a highly effective combination of modulators of the CFTR protein (Cystic fibrosis transmembrane conductance regulator), for the treatment of cystic fibrosis (CF) in patients with at least one F508del mutation of the CFTR gene (the most common mutation in CF). The active substances are elexacaftor, tezacaftor and ivacaftor. Currently, more than 85% of people with CF are eligible for CFTR modulator therapy, which has been shown to improve respiratory function and quality of life and reduce pulmonary exacerbations.
Key Words: cystic fibrosis, modulators of the CFTR, elexacaftor, tezacaftor, ivacaftor
Mukoviszidose
Mukoviszidose ist eine potenziell schwere monogenetische Erkrankung, die autosomal-rezessiv vererbt wird und weltweit mehr als 100’000 Menschen betrifft (1, 2). Sie wird durch Mutationen im CFTR-Gen verursacht, die zu einer veränderten Synthese oder Funktion des CFTR-Proteins (Cystic Fibrosis Transmembrane conductance Regulator, im Folgenden pCFTR) führen. Seit der ersten Entdeckung der häufigsten Mutation (F508del) wurden über 2000 Varianten des CFTR-Gens beschrieben (1, 2), von denen etwa 400 als mit Mukoviszidose assoziiert gelten. Sie werden in die Klassen I bis VI eingeteilt, je nachdem, wie sie sich auf die Produktion und Funktion des pCFTR auswirken (1, 2). Das pCFTR ist ein transmembraner Chloridkanal, der sich auf der apikalen Seite sekretorischer Epithelien, insbesondere der Schweissdrüsen, der Atemwege, des Gastrointestinaltrakts, der Bauchspeicheldrüse und der Samenleiter befindet (1, 2). Es reguliert den Salz- und Wasserhaushalt auf der Zelloberfläche und eine Fehlfunktion dieses Proteins führt zu zähflüssigem Schleim und Sekreten. Die klinischen Manifestationen der Krankheit sind multisystemisch und treten je nach pCFTR-Dysfunktion in unterschiedlichem Alter auf (3, 4).
In der Lunge ist die Mukoviszidose durch eine schwere Störung der mukoziliären Clearance, eine Entzündung und eine chronische bakterielle Kolonisierung der oberen und unteren Atemwege gekennzeichnet, die zu Bronchiektasien und einem allmählichen Rückgang der Lungenfunktion führt. Eine exokrine Pankreasinsuffizienz mit Malabsorption von Lipiden und fettlöslichen Vitaminen ist sehr häufig, ebenso wie Verstopfung. Mukoviszidose-bedingter Diabetes tritt bei 30 % der Patienten über 18 Jahren auf und die Prävalenz steigt mit zunehmendem Alter, während die biliäre Zirrhose viel seltener ist (3, 4). Die Behandlung der Mukoviszidose basierte lange Zeit auf einer symptomatischen Behandlung, die zwar anstrengend war, aber die Lebenserwartung der Patienten erheblich verlängerte. Seit den 2010er-Jahren steht eine neue Klasse von Medikamenten zur Verfügung, die pCFTR-Modulatoren, mit denen die Aktivität des pCFTR wiederhergestellt werden kann (1).
Modulatoren des CFTR-Proteins
Im Gegensatz zu symptomatischen Therapien, die sich auf die Behandlung von Komplikationen der Mukoviszidose konzentrieren, sind pCFTR-Modulatoren kleine Moleküle, die am Ursprung des Problems ansetzen (Abb. 1).
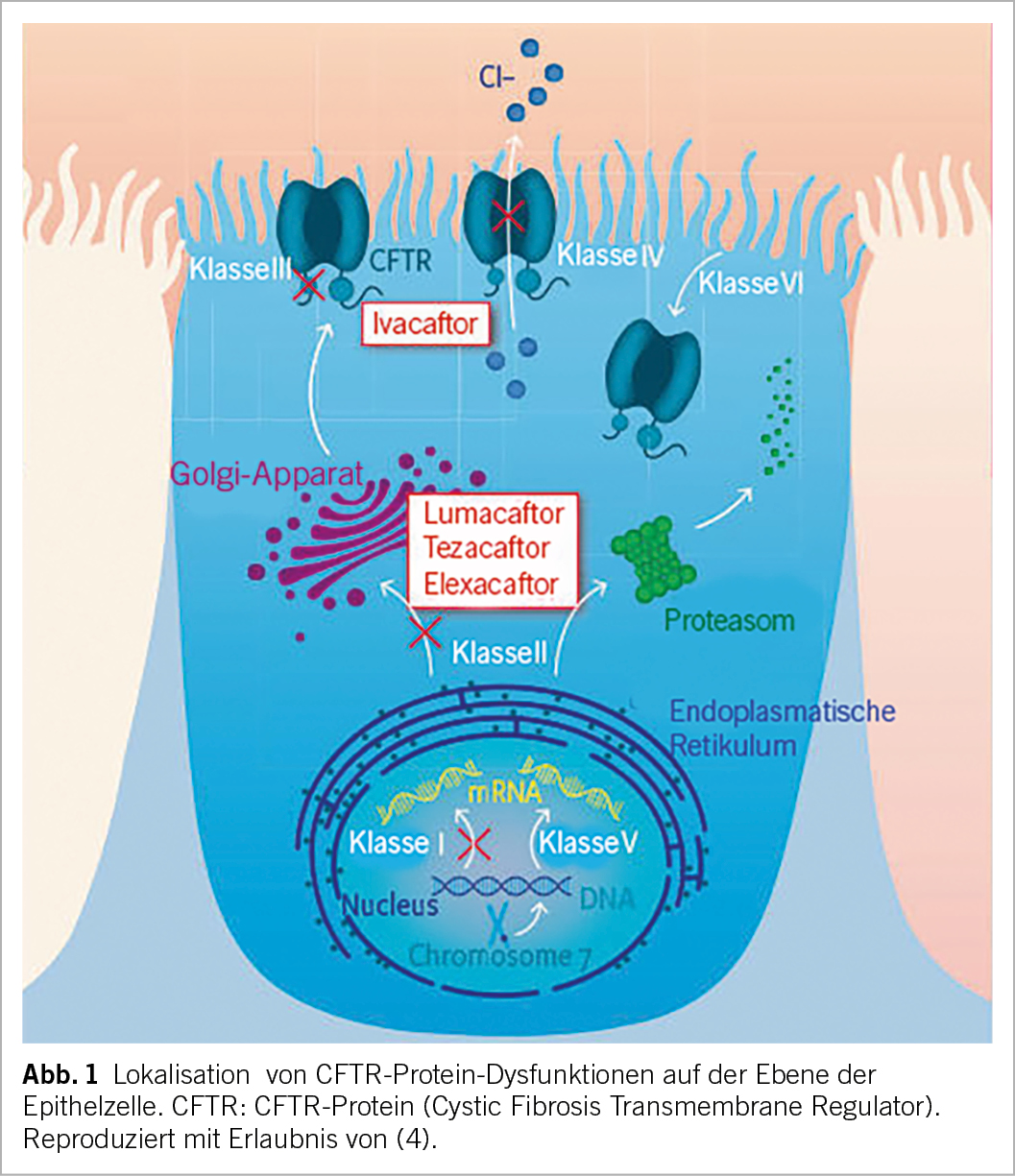
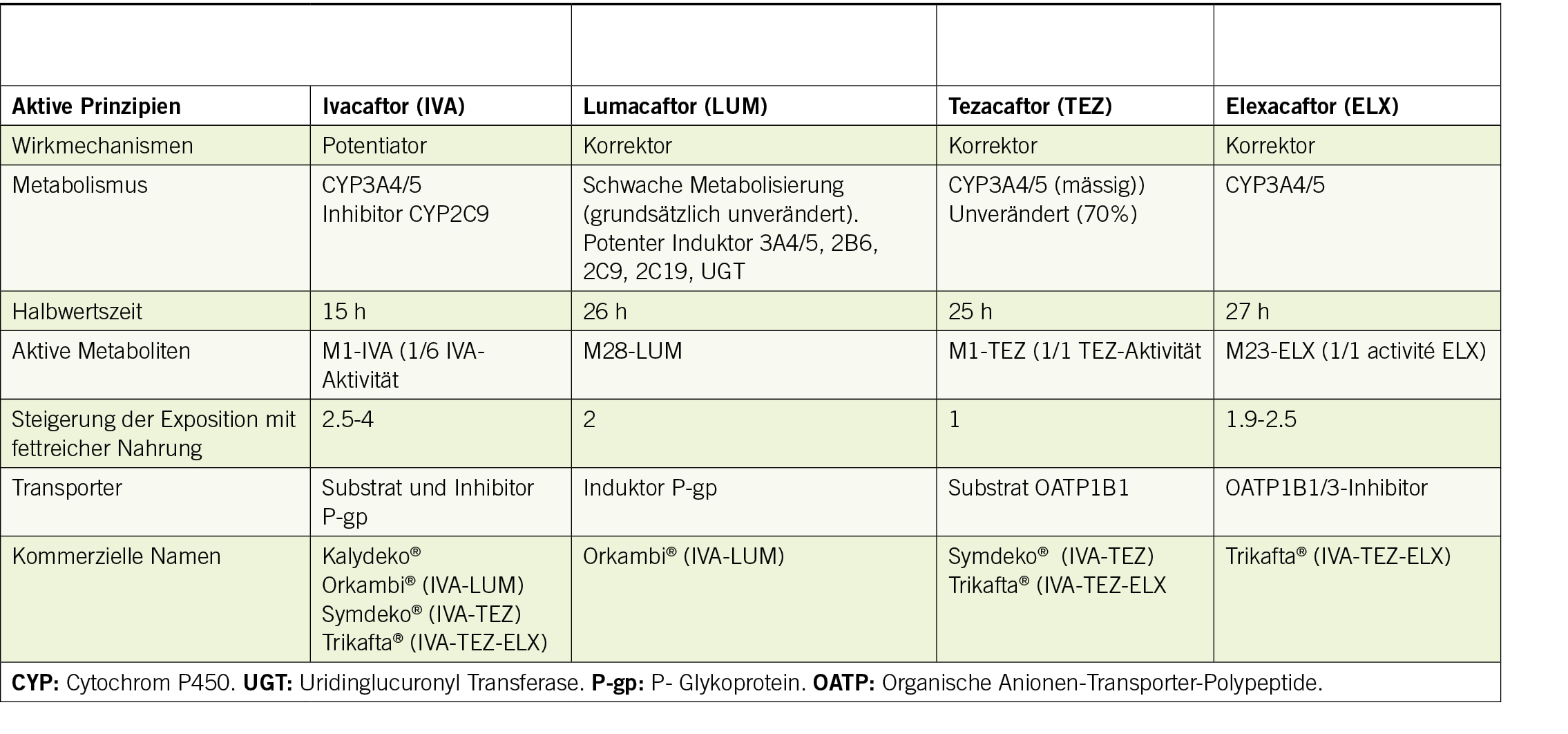
Es gibt zwei Arten von Modulatoren: Potentiatoren (Ivacaftor) verbessern die Funktion des pCFTR, indem sie die Öffnungszeit der Chloridkanäle begünstigen. Die Korrektoren (Lumacaftor, Tezacaftor und Elexacaftor) stabilisieren den pCFTR und erleichtern seinen Transport zur Zellmembran (1). Derzeit sind vier Medikamente als Mono-, Zwei- oder Dreifachtherapie auf dem Markt, von denen einige Merkmale in Tabelle 1 beschrieben sind.
Wirksamkeit und Sicherheit von Modulatoren
Im Jahr 2019 wurden zwei pivotale Studien zur Dreifachtherapie mit Elexacaftor/Tezacaftor/Ivacaftor (Trikafta®) bei 113 homozygoten F508del-Patienten (5) und 403 heterozygoten F508del/Varianten-Kompositpatienten mit minimaler Funktion, die durch die bisherigen Modulatorkombinationen nicht korrigiert werden konnte, veröffentlicht (6). Die erste Studie umfasste nur 4 Wochen und die Kontrollgruppe bestand aus Patienten, die mit Ivacaftor/Tezacaftor behandelt wurden. Die Zugabe von Elexacaftor führte zu einer zusätzlichen Steigerung des maximalen exspiratorischen Einsekundenvolumens (FEV1) um 10,0% und einer Senkung des Schweisschlorids um 45 mmol/l (p < 0,0001). Die zweite Studie führte nach 24 Wochen zu einer Verbesserung des FEV1 um 13,9%, einem Rückgang der Rate an Lungenexazerbationen um 63%, einer Verbesserung des CFQ-R-Scores (Lebensqualitätsscore) um 20,2 Punkte und einer Senkung des Chlorids im Schweiss um 42 mmol/l (p<0,001 für alle Vergleiche). Diese Ergebnisse wurden durch prospektive Real-Life-Studien bestätigt, die auch dazu führten, dass die FDA die Zulassung auf Kinder ab 6 Jahren und vor kurzem auf Kinder ab 2 Jahren ausdehnte (7, 8).
Aus Sicht der Toxizität sind die Modulatoren relativ gut verträglich (9-11). Die Kombination Ivacaftor/Lumacaftor weist jedoch höhere Raten an respiratorischen Nebenwirkungen auf, die offenbar auf Lumacaftor zurückzuführen sind (10). Ein vorübergehender Anstieg von Husten/Bronchorrhoe innerhalb von 48 Stunden nach Behandlungsbeginn ist die Folge einer verbesserten Sekretclearance und nimmt danach wieder ab. Andere Nebenwirkungen treten vor allem im Magen-Darm-Bereich (Durchfall, Übelkeit, Bauchschmerzen) und bei der Leber (Anstieg der Transaminasen) auf (9-11). Allergien vom Typ Ausschlag oder Überempfindlichkeit wurden selten berichtet. Ein Signal für eine Beeinträchtigung der psychischen Gesundheit (einschliesslich Depressionen oder neuropsychiatrische Störungen) wurde auf der Grundlage einiger Kohortenstudien und Fallberichte identifiziert (9), obwohl ein kausaler Zusammenhang mit CFTR-Modulatoren nicht formell nachgewiesen werden konnte.
Pharmakokinetische Erwägungen
Die pCFTR-Modulatoren werden hauptsächlich über die Zytochrome (CYP) 3A4/5 eliminiert (Tab. 1). Ivacaftor und Elexacaftor können auch Substrate, Inhibitoren oder Induktoren von P-gp und/oder OATP1B1/3 sein. Daher stellen sie ein erhebliches Risiko für Arzneimittelwechselwirkungen dar, insbesondere mit Induktoren oder Inhibitoren von CYP3A4/5. Eine deutliche Verringerung der Exposition (89%) und ein 15-facher Anstieg der Ivacaftor-Konzentrationen bei gleichzeitiger Verabreichung mit Rifampicin bzw. Itraconazol wurden beobachtet (11). Geringere, aber signifikante Veränderungen werden bei moderaten Inhibitoren oder Induktoren von CYP3A4/5 (z.B. Rifabutin oder Fluconazol) erwartet. Obwohl keine Arzneimittelwechselwirkungen zwischen Trikafta® und OATP1B1/3-Induktoren berichtet wurden, könnten die OATP1B1/3-Inhibitoren Gemfibrozil und Ciclosporin theoretisch zu einem Anstieg der Serumkonzentrationen von Trikafta® führen (12). Da Ivacaftor als CYP2C9-Hemmer die Wirkung bestimmter Arzneimittel, die ein Substrat dieses Enzyms darstellen (Acenocoumarol, Glibenclamid), verstärken kann, ist bei der Verabreichung von Arzneimitteln, die ein Substrat von OATP1B1 darstellen, wie z. B. Statine, Glibenclamid oder Repaglinid, Vorsicht geboten und eine angemessene Überwachung erforderlich. Da Bilirubin ein Substrat von OATP1B1 und OATP1B3 ist, kann es zu leichten Erhöhungen des durchschnittlichen Gesamtbilirubinspiegels kommen (10, 11).
Bei Patienten, die mit Trikafta® behandelt werden, wurde über eine erhebliche Variabilität der Plasmakonzentrationen berichtet, die mit bestimmten Faktoren in Zusammenhang steht, wie Alter und Gewicht bei Kindern, fettreiche Nahrung, die die Exposition gegenüber diesen Arzneimitteln signifikant erhöht, Ko-Medikationen mit dem Risiko von Wechselwirkungen und Veränderungen der Elimination bei Leber- oder Niereninsuffizienz. Die Auswirkungen dieser pharmakokinetischen Veränderungen auf das Ansprechen und die Verträglichkeit dieser Medikamente sind jedoch noch weitgehend unbekannt. Die Überwachung der Therapie durch die Messung der Plasmaspiegel ist potenziell hilfreich, um die Dosierung dieser Medikamente anzupassen (13).
Was ist in der Praxis zu beachten?
Die Dosen von Trikafta® sollten im Abstand von ca. 12 Stunden zu einer fettreichen Mahlzeit eingenommen werden. Bei mässiger Leberinsuffizienz sollte die Dosierung reduziert werden (11).
Die gleichzeitige Verschreibung von starken CYP3A4/5-Induktoren ist aufgrund des Risikos eines Wirkungsverlustes kontraindiziert. Bei starken und schwachen CYP3A4/5-Inhibitoren wird eine Dosisreduktion empfohlen, um das Risiko von Nebenwirkungen zu verringern. Vorsicht und eine angemessene Überwachung sind geboten, wenn Arzneimittel, die Substrate von CYP2C9, P-gp, OATP1B1 und OATP1B3 sind, zusammen mit Trikafta® verabreicht werden, da die Exposition gegenüber diesen Arzneimitteln erhöht sein kann.
Es wird empfohlen, die Transaminasen (ALAT und ASAT) und das Gesamtbilirubin bei allen Patienten vor Beginn der Behandlung, im ersten Jahr alle drei Monate und danach mindestens einmal jährlich zu kontrollieren (11). Aufgrund des Risikos einer schweren Verschlechterung der Atmung nach Absetzen der Behandlung ist eine Unterstützung der Therapietreue und der Kenntnis der Herausforderungen durch den Patienten und das Pflegepersonal erforderlich (14).
Die Kosten für eine Packung Trikafta® mit 84 Tabletten (28 Tage) belaufen sich auf CHF 17’516.15 und erfordern die Zustimmung der Versicherer zur Kostenübernahme mit einer vorherigen Beurteilung durch den Vertrauensarzt der Versicherung.
Schlussfolgerung
Die pCFTR-Modulatoren haben die Behandlung von Patienten mit Mukoviszidose revolutioniert und zu einer deutlichen Verbesserung der Lungenfunktion und der Lebensqualität sowie zu einer Verringerung des Risikos von Lungenexazerbationen geführt. Wie bei jeder neuen Behandlung sind jedoch die langfristige Wirksamkeit, insbesondere die extrapulmonale Wirksamkeit, und die langfristigen Nebenwirkungen noch unbekannt und es müssen bestimmte Vorsichtsmassnahmen ergriffen werden, um das Risiko einer Toxizität zu minimieren. Weitere Wirkstoffe werden derzeit untersucht und sollen in den nächsten Jahren auf den Markt kommen (15).
Copyright Aerzteverlag medinfo AG
Service de pharmacie, Centre Hospitalier Universitaire Vaudois et
Université de Lausanne
Lausanne
ermindo.di-paolo@chuv.ch
Unité de mucoviscidose adulte, Service de Pneumologie,
Centre Hospitalier Universitaire Vaudois et Université de Lausanne
Lausanne
georgia.mitropoulou@chuv.ch
Centre de Recherche et d’ Innovation en Sciences Pharmaceutiques
cliniques Centre Hospitalier Universitaire et Université de Lausanne
Suisse Rue du Bugnon 19
1011 Lausanne
Chantal.Csajka@chuv.ch
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel angegeben.
1. Shteinberg M, Haq IJ, Polineni D, et al. Cystic fibrosis. Lancet 2021;397:2195-2211.
2. Bell SC, Mall MA, Gutierrez H, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med 2020; 8:65-124.
3. Koutsokera A, Sauty A et al. Swiss recommendations for adult cystic fibrosis care. https://www.revmed.ch/guidelines/swiss-recommendations-for-adult-cystic-fibrosis-care (letzter Zugriff 20.02.2023).
4. Sauty A, Plojoux J, Mornand A, et al. Révolution dans le traitement de la mucoviscidose. Rev Med Suisse 2020; 16:1229-35.
5. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019;394:1940-8.
6. Middleton PG, Mall MA, Drevínek P, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med 2019;381:1809-19.
7. Trikafta® Swiss Public Assessment Report. Swissmedicinfo. 30 March 2022.
8. Nichols DP, Paynter AC, Heltshe SL, et al. Clinical effectiveness of Elexacaftor/Tezacaftor/Ivacaftor in people with cystic fibrosis: a clinical trial. Am J Respir Crit Care Med 2022;205:529-39.
9. Dagenais RVE, Su VCH, Quon BS. Real-world safety of CFTR modulators in the treatment of cystic fibrosis: a systematic review. J Clin Med 2020;10:23. doi: 10.3390/jcm10010023.
10. Gramegna A, Contarini M, Aliberti S, et al. From ivacaftor to triple combination: a systematic review of efficacy and safety of CFTR modulators in people with cystic fibrosis. Int J Mol Sci 2020 ;21:5882. doi: 10.3390/ijms21165882.
11. https://www.swissmedicinfo.ch, Fachinformation Trikafta® (letzter Zugriff 20.02.2023).
12. Purkayastha D, Agtarap K, Wong K, et al. Drug-drug interactions with CFTR modulator therapy in cystic fibrosis: focus on Trikafta®/Kaftrio®. J Cyst Fibros 2023. doi: 10.1016/j.jcf.2023.01.005.
13. Choong E, Sauty A, Koutsokera A, et al. Therapeutic drug monitoring of ivacaftor, lumacaftor, tezacaftor, and elexacaftor in cystic fibrosis: where are we now? Pharmaceutics 2022;14:1674. doi: 10.3390/pharmaceutics14081674.
14. Mitropoulou G, Balmpouzis Z, Plojoux, J, et al. Effects of elexacaftor−tezacaftor−ivacaftor discontinuation in cystic fibrosis. Respir Med Res 2022;82:100972. doi: 10.1016/j.resmer.2022.100972.
15. https://apps.cff.org/trials/pipeline (letzter Zugriff 20.02.2023).



der informierte @rzt
- Vol. 13
- Ausgabe 7
- Juli 2023