Klassifikation, Pathomechanismen und klinischer Verlauf
Interstitielle Pneumopathie ist ein Überbegriff für eine heterogene Gruppe von über 200 verschiedenen Lungenerkrankungen, deren pathologischen Prozesse sich hauptsächlich im Interstitium abspielen. Sie führen bei den Patient/-innen zu einer verminderten Aufnahme von Sauerstoff, konsekutiver Atemnot, gefolgt von einer Verschlechterung der Lebensqualität und enden teilweise in Lungenversagen und Tod [1, 2]. Es sind grundsätzlich seltene Erkrankungen mit jedoch grosser Variabilität der Inzidenzen von zum Beispiel 0.3 pro 100‘000 Personen pro Jahr für die Lymphangioleiomyomatose respektive 5-9 pro 100‘000 Personen pro Jahr für die Idiopathische Pulmonale Fibrose (IPF) [3]. Generell sind Inzidenz und Prävalenz einerseits beeinflusst durch geografische Faktoren, aber auch durch Patienten-bedingte Faktoren wie Ethnie, Alter, Geschlecht und genetische Prädisposition [4].
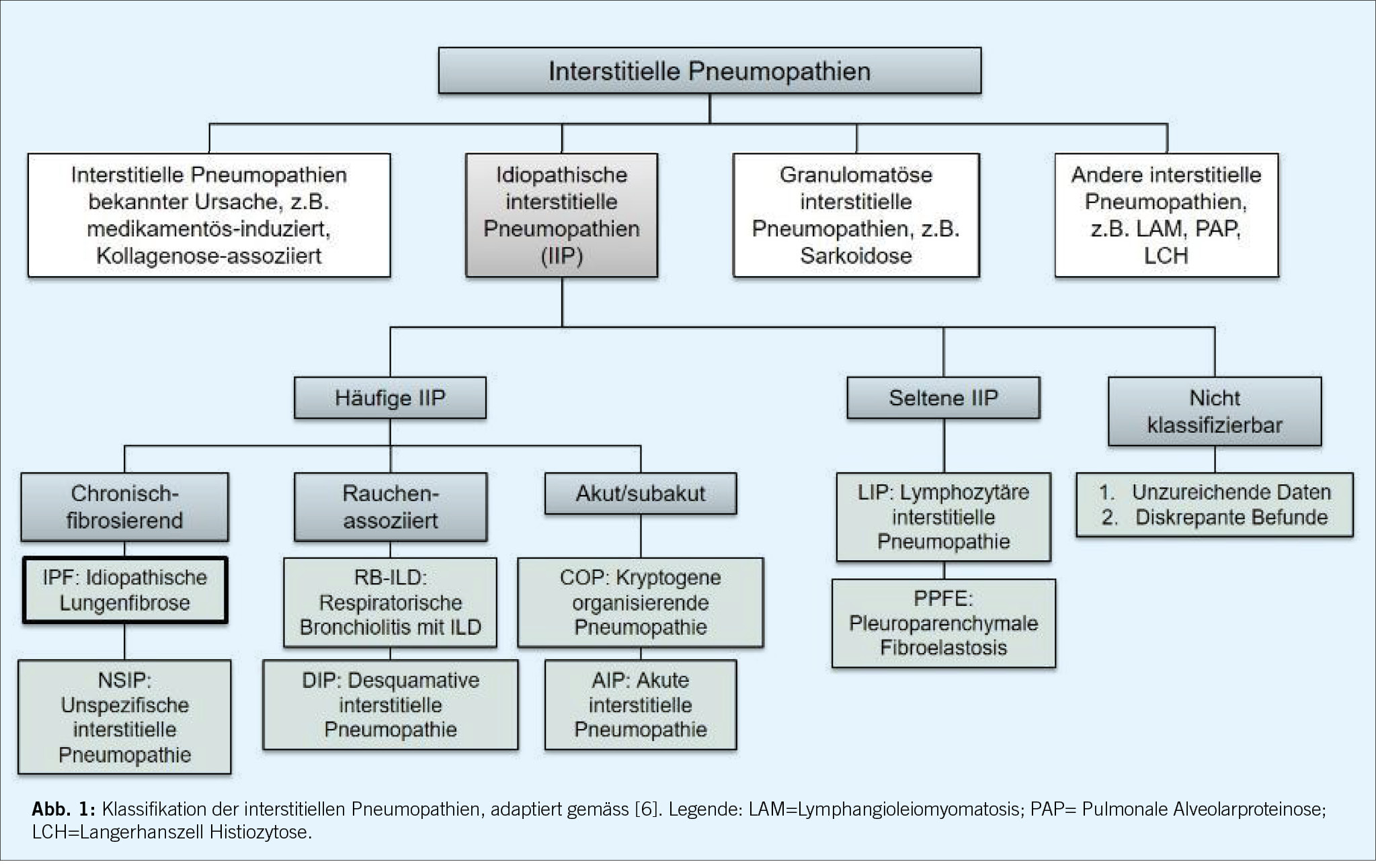
Bereits im 19. Jahrhundert wurden durch die Pathologen Georg Eduard von Rindfleisch und David Paul von Hansemann fibrosierende interstitielle Lungenerkrankungen beschrieben. 1935 beobachteten Louis Hamman und Arnold Rich vom Johns Hopkins Hospital mehrere Fälle von akuten und schweren Verläufen bei Patient/-innen mit Lungenfibrose, und wurden damit Namensgeber des “Hamman-Rich-Syndroms”. Eine erste Klassifikation der interstitiellen Pneumopathien entstand in den 60-er Jahren des letzten Jahrhunderts [5], und diese wurde seither mehrfach überarbeitet und durch neu beschriebene Entitäten erweitert. Die letzte offizielle Klassifikation, welche von Expertinnen und Experten im Auftrag der Amerikanischen und Europäischen Gesellschaften für Pneumologie erarbeitet worden ist, wurde 2013 publiziert und gliedert die interstitiellen Pneumopathien in vier Hauptgruppen: 1. Interstitielle Pneumopathien bekannter Ursachen, 2. idiopathische interstitielle Pneumopathien, 3. granulomatöse interstitielle Pneumopathien und 4. andere Entitäten (Abbildung 1) [6]. Zu den interstitiellen Pneumopathien mit bekannter Ursache gehören unter anderem die Konnektivitiden-assoziierten Erkrankungen, die Pneumokoniosen (ausgelöst durch die meist beruflich bedingte Inhalation anorganischer Stäube), oder die medikamentös-induzierten Pneumopathien. Die idiopathischen interstitiellen Pneumopathien (IIP) werden weiter aufgeteilt in häufige, seltene und nicht klassifizierbare Erkrankungen, wobei die erst 2004 erstmals beschriebenen Pleuroparenchymale Fibroelastosis ein Beispiel einer neu in die Klassifikation aufgenommenen seltenen IIP ist [7]. Die idiopathische pulmonale Fibrose (IPF) ist sowohl die häufigste IIP als auch diejenige interstitielle Pneumopathie mit dem ungünstigsten Krankheitsverlauf, aber auch jene, für welche bezüglich Therapie am meisten Evidenz vorliegt.
Die Ursachen und pathophysiologische Mechanismen der verschiedenen Pneumopathien sind sehr unterschiedlich, mit überwiegend entzündlichen Prozessen, wie zum Beispiel die Sarkoidose, Zigarettenrauch-assoziierte Erkrankungen, wie die Respiratorische Bronchiolitis mit interstitieller Pneumopathie und die desquamative interstitielle Pneumopathie, oder aber vorwiegend fibrotischer Pneumopathien, wie die IPF.
Entsprechend sind auch die Krankheitsverläufe sehr unterschiedlich mit einerseits vollständiger Reversibilität, typischerweise bei der organisierenden Pneumonie zu erwarten, und andererseits progredienter Verschlechterung mit irreversibler Fibrosierung, wie bei der IPF [4]. Grundsätzlich ist das Ausmass der Fibrosierung respektive der Anteil an Inflammation von Bedeutung, wobei eine zunehmende Vernarbung/Fibrosierung mit einem ungünstigen Verlauf assoziiert ist, hingegen dominant entzündliche Veränderungen ein gutes Ansprechen auf Kortikosteroide erwarten lassen. Nicht selten sind interstitielle Pneumopathien, die primär eine rein entzündliche Komponente aufweisen und erst im weiteren, längerfristigen Verlauf in eine irreversible Fibrosierung münden. Bei diesen Patient/-innen, welche trotz etablierter Therapie eine klinische, funktionelle oder radiologische Verschlechterung der interstitiellen Pneumopathie erleiden, sollte die Diagnose einer Progressiven Pulmonalen Fibrose (PPF) gestellt werden [2].
Diagnostik
Die Heterogenität der interstitiellen Pneumopathien lässt die Diagnosestellung teilweise zur langwierigen und komplexen Detektivarbeit werden. Die ausführliche Anamnese einschliesslich der Fragen nach beruflicher oder privater inhalativer Schadstoffexposition, Medikamenten-Anamnese und einer gezielten Familienanamnese bildet den Ausgangspunkt im diagnostischen Prozedere. Die körperliche Untersuchung dient unter anderem der Suche nach Zeichen einer bisher nicht bekannten zugrundeliegenden Systemerkrankung. Diese Suche wird erweitert durch die Bestimmung von Autoantikörpern bzgl. Konnektivitide resp. Rheumatoider Arthritis und findet idealerweise in enger interdisziplinärer Zusammenarbeit mit den Kolleg/-innen der Rheumatologie statt. Die Lungenfunktionsdiagnostik dient der Bestimmung der Lungenvolumina sowie der Messung der Diffusionskapazität, wenn möglich ergänzt durch einen Belastungstest (Spiroergometrie oder 6-Minutengehtest). Die Resultate der Lungenfunktion liefern Informationen bezüglich des Schweregrads der Erkrankung, dienen der Beurteilung des Therapieansprechens und der Abschätzung der Prognose [4].
Eine weitere zentrale Rolle spielt die Computertomographie der Lungen, wobei die Befundung und Zuordnung der verschiedenen radiologischen Muster idealerweise durch erfahrene Thorax-Radiolog/-innen erfolgen sollte. Das CT-radiologische Muster bestimmt das weitere diagnostische Prozedere, wobei dieses im Rahmen eines interdisziplinären Boards festgelegt werden sollte. Eine broncho-alveoläre Lavage mittels flexibler Bronchoskopie kann insbesondere bei alveolären Prozessen, wie der pulmonale Alveolarproteinose oder der eosinophilen Pneumonie, bereits diagnostisch sein. Die Histologie mittels transbronchialer Zangenbiopsie ist bei Erkrankungen wie der Organisierenden Pneumonie oftmals diagnostisch, wohingegen bei subpleural dominierenden Veränderungen eine transbronchialer Kryobiopsie empfohlen wird [8]. Nur noch selten muss zur Diagnosesicherung eine chirurgische Lungenbiopsie durchgeführt werden. Generell sollte die klinische Relevanz der durch die Intervention gewonnenen Information gegen das Interventionsrisiko abgewogen werden. Schliesslich sollten alle erhobenen Befunde – im Sinne einer Konsensus-Diagnose – in einem interdisziplinären Board, bestehend aus Pneumolog/-innen, Radiolog/-innen, Patholog/-innen und Rheumatolog/-innen, diskutiert werden. Trotz ausführlicher Diagnostik kann bei ca. 10-20% aller Patient/-innen jedoch keine sichere Klassifizierung der interstitiellen Pneumopathie vorgenommen werden, so dass man sich mit der Diagnose „unklassifizierbare interstitielle Pneumopathie“ begnügen muss [9].
Therapie
Die Wahl der medikamentösen Therapie richtet sich grundsätzlich nach der zugrundeliegenden Ursache der interstitiellen Pneumopathie [10]. Eine primär anti-inflammatorische Behandlung mit Kortikosteroiden oder Immunsuppressiva ist bei Erkrankungen mit dominant entzündlicher Komponente vielversprechend, obwohl diesbezüglich wenig Evidenz vorliegt; die Sarkoidose, die cryptogen organisierende Pneumonie und auch die eosinophile Pneumonie sind Beispiele dafür. Bei prädominant fibrosierenden interstitiellen Pneumopathien sind anti-entzündliche Substanzen meist wirkungslos, oder – wie bei der IPF- sogar kontraindiziert.
Erst vor wenigen Jahren konnten positive Studiendaten für zwei anti-fibrotisch wirksame Medikamente (Nintedanib und Pirfenidon) publiziert werden, wobei in der Schweiz aktuell für die Therapie der IPF sowohl Nintedanib als auch Pirfenidon und bei PPF nur Nintedanib zugelassen sind. Für beide Wirkstoffe konnte lediglich eine Verlangsamung der Krankheitsprogression gezeigt werden [11, 12] [13].
Ausser bei der IPF – welche immer progressiv fibrosierend verläuft und deshalb immer und ausschliesslich anti-fibrotisch behandelt werden muss – kann die Unterscheidung zwischen prädominant entzündlich versus prädominant fibrotisch sehr herausfordernd sein; die Entscheidung stützt sich auf die zugrundeliegende Diagnose, zytologische (broncho-alveoläre Lavage) und histologische (Lungenbiopsie) Befunde und das CT-radiologische Muster [9].
Nicht-medikamentöse Therapien respektive Angebote umfassen die Sauerstoff-Supplementation, ambulante oder stationäre pulmonale Rehabilitation, psychologische Begleitung und palliativmedizinische Betreuung [14]. Dieses multimodale und interprofessionelle Betreuungskonzept ist insbesondere für Patient/-innen mit progressiv fibrosierendem Krankheitsverlauf relevant und sollte im Sinne einer gesundheitlichen Vorausplanung möglichst früh in der klinischen Betreuung berücksichtigt werden.
Ausblick
Nach grossen Fortschritten im Verständnis der Pathomechanismen bei IPF und PPF und darauf basierenden neuen Therapieansätzen bleibt die Grundlagen- und klinische Forschung im Bereich der interstitiellen Pneumopathien weiter sehr aktiv und innovativ. Von neuen diagnostischen Ansätzen erhofft man sich insbesondere eine möglichst frühe Diagnose bei gleichzeitig geringer Invasivität. Blut, Bio-
marker, molekulare Marker, neue bildgeberische Verfahren, aber auch KI-unterstützte Bild-Analysen sind vielversprechende Instrumente [15-19].
Von therapeutischer Seite konnten für zwei neue Substanzen in Phase 2 Studien sehr vielversprechende Daten gezeigt werden: BI 1015550, ein oraler Phosphodiesterase 4B Inhibitor, verhinderte eine Verschlechterung der Lungenfunktion bei IPF Patient/-innen [20], und auch für BMS-986278, ein oraler Antagonist von LPA1 (=lysophosphatidic acid receptor 1), ergaben sich positive Resultate bezüglich Krankheitsprogression (NCT04308681). Für beide Substanzen laufen aktuell Phase 3 Studien, deren Resultate mit grossem Interesse erwartet werden.
Das Feld der interstitiellen Pneumopathien bleibt somit interessant, vielseitig und auch herausfordernd, und wir sind sowohl in der Diagnostik als auch in der langfristigen Betreuung unserer Patient/-innen auf eine gute interdisziplinäre und interprofessionelle Zusammenarbeit angewiesen.
Prof. Dr. Dr. Katrin Hostettler, katrin.hostettler@usb.ch
Kaderärztin
Klinik für Pneumologie & Departement Biomedizin
Universitätsspital Basel
Petersgraben 4
CH-4031 Basel
Klinik für Pneumologie & Departement Biomedizin
Universitätsspital Basel
Petersgraben 4
CH-4031 Basel
Non-conditional grants von Boehringer-Ingelheim, Schweiz; Non-conditional grants von Roche Pharma, Schweiz; Speaker-fees von Boehringer-Ingelheim, Schweiz.
Literatur:
1. Kim, H.J., D. Perlman, and R. Tomic, Natural history of idiopathic pulmonary fibrosis. Respir Med, 2015. 109(6): p. 661-70.
2. Raghu, G., et al., Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med, 2022. 205(9): p. e18-e47.
3. Shah Gupta, R., et al., Incidence and prevalence of interstitial lung diseases worldwide: a systematic literature review. BMJ Open Respir Res, 2023. 10(1).
4. Koudstaal, T., et al., Pulmonary fibrosis: from pathogenesis to clinical decision-making. Trends Mol Med, 2023. 29(12): p. 1076-1087.
5. Liebow AA, C.C., The interstitial pneumonias, in Frontiers of pulmonary radiology, P.E. Simon M, LeMay M, Editor. 1969, Grune & Stratton: New York p. 102–141.
6. Travis, W.D., et al., An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med, 2013. 188(6): p. 733-48.
7. Frankel, S.K., et al., Idiopathic pleuroparenchymal fibroelastosis: description of a novel clinicopathologic entity. Chest, 2004. 126(6): p. 2007-13.
8. Korevaar, D.A., et al., European Respiratory Society guidelines on transbronchial lung cryobiopsy in the diagnosis of interstitial lung diseases. Eur Respir J, 2022. 60(5).
9. Ryerson, C.J., et al., A contemporary practical approach to the multidisciplinary management of unclassifiable interstitial lung disease. Eur Respir J, 2021. 58(6).
10. Wijsenbeek, M. and V. Cottin, Spectrum of Fibrotic Lung Diseases. N Engl J Med, 2020. 383(10): p. 958-968.
11. King, T.E., Jr., et al., A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med, 2014. 370(22): p. 2083-92.
12. Richeldi, L., et al., Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med, 2014. 370(22): p. 2071-82.
13. Flaherty, K.R., et al., Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N Engl J Med, 2019. 381(18): p. 1718-1727.
14. Kreuter, M., et al., Palliative care in interstitial lung disease: living well. Lancet Respir Med, 2017. 5(12): p. 968-980.
15. Kreuter, M., et al., Monocyte Count as a Prognostic Biomarker in Patients with Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med, 2021. 204(1): p. 74-81.
16. Stainer, A., et al., Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. Int J Mol Sci, 2021. 22(12).
17. Inoue, Y., et al., Diagnostic and Prognostic Biomarkers for Chronic Fibrosing Interstitial Lung Diseases With a Progressive Phenotype. Chest, 2020. 158(2): p. 646-659.
18. Röhrich, M., et al., Fibroblast Activation Protein-Specific PET/CT Imaging in Fibrotic Interstitial Lung Diseases and Lung Cancer: A Translational Exploratory Study. J Nucl Med, 2022. 63(1): p. 127-133.
19. Walsh, S.L.F., et al., Deep Learning-based Outcome Prediction in Progressive Fibrotic Lung Disease Using High-Resolution Computed Tomography. Am J Respir Crit Care Med, 2022. 206(7): p. 883-891.
20. Richeldi, L., et al., Trial of a Preferential Phosphodiesterase 4B Inhibitor for Idiopathic Pulmonary Fibrosis. N Engl J Med, 2022. 386(23): p. 2178-2187.
Therapeutische Umschau
- Vol. 81
- Ausgabe 1
- Februar 2024