

La sclérose en plaques est la maladie du SNC à médiation auto-immune la plus fréquente et touche surtout les jeunes femmes. Elle se caractérise par une évolution par poussées. Les poussées peuvent se manifester de manière variable, les premiers symptômes fréquents étant une névrite optique ou des troubles de la sensibilité. L’IRM du neuroaxe révèle des lésions démyélinisantes réparties en foyers, qui constituent un corollaire à l’imagerie morphologique. Outre le traitement aigu en cas de poussée, il existe aujourd’hui de nombreuses préparations modifiant l’évolution de la maladie, qui permettent d’influencer de manière significative le cours de la maladie.
Multiple sclerosis is the most common autoimmune-mediated CNS disease and primarily affects young women. A relapsing-remitting course of the disease is characteristic. Relapse symptoms are highly variable, common initial symptoms are optic neuritis or sensory disturbances. Focally distributed demyelinating lesions are typical findings in the MRI of the neuroaxis. In addition to acute treatment options in case of a relapse, numerous disease-modifying drugs with significant impact on disease progression have become available.
Key Words: Multiple sclerosis, McDonald criteria, immune modulation
Vignette de cas
Une étudiante en psychologie de 24 ans se présente à votre cabinet de médecine générale en raison d’une maladresse de la main droite qui dure depuis environ trois semaines. Au début, elle ne l’a remarquée que lorsqu’elle jouait au piano, mais la semaine dernière, un verre lui a presque glissé des mains à deux reprises, si bien qu’elle s’inquiète de plus en plus. Au contact, elle décrit une sensation de fourrure modifiée dans la main. Quelles sont les mesures que vous prenez et quelle pourrait être la cause de ces symptômes?
Les généralités
La sclérose en plaques (SEP) est l’une des maladies auto-immunes les plus fréquentes chez les jeunes et la plus fréquente des maladies inflammatoires chroniques du SNC à médiation auto-immune. Rien qu’en Suisse, environ 15 000 personnes sont touchées (1). La maladie se caractérise par une évolution généralement par poussées et un schéma lésionnel en forme de foyer à l’imagerie par résonance magnétique. La sclérose en plaques est plus fréquente chez les femmes que chez les hommes (sex-ratio 3:1). Dans 80% des cas, elle se manifeste pour la première fois chez le jeune adulte, entre 20 et 40 ans (2). L’étiologie n’est qu’incomplètement comprise à ce jour. Outre l’autoréactivité immunologique, une prédisposition génétique ainsi que des facteurs environnementaux (carence en vitamine D, obésité, tabagisme) sont discutés en cas d’accumulation familiale. Une infection par le virus d’Epstein Barr peut augmenter considérablement le risque de développer une SEP. D’un point de vue physiopathologique, la démyélinisation inflammatoire des fibres nerveuses est la première cause de dommages axonaux secondaires.
La clinique
Le tableau clinique est très variable, en fonction de la localisation de la lésion. Les premiers symptômes typiques de la poussée, qui doivent faire penser à une sclérose en plaques dans le cadre du diagnostic différentiel, sont une détérioration unilatérale de l’acuité visuelle, le plus souvent dans le sens d’un trouble de la vision des couleurs accompagné d’une douleur des mouvements oculaires dans le cadre d’une névrite optique, et des déficits sensitifs ou moteurs (unilatéraux ou en tant que symptômes transversaux) avec ou sans troubles de la fonction vésicale. Les troubles de la sensibilité se manifestent souvent sous forme de paresthésies ou de dysesthésies sous forme de fourrure, de fourmillements, de sensation de ceinture ou de corset avec une répartition asymétrique et plutôt distale. En outre, des troubles de l’équilibre et de la coordination, une vision double et des vertiges sont fréquents comme corrélats d’une atteinte du tronc cérébral. Une poussée est définie comme un symptôme nouvellement apparu qui ne peut pas être expliqué dans le cadre d’une infection ou d’une augmentation de la température corporelle, appelé phénomène d’Uhthoff. La durée minimale est de 24 heures et un intervalle minimum de 30 jours est appliqué par rapport au dernier événement de poussée. Contrairement à d’autres diagnostics différentiels neurologiques (accident vasculaire cérébral, crise d’épilepsie), les symptômes s’installent généralement lentement, sur plusieurs heures ou jours. L’intensité est également très variable. Elle va de troubles discrets de la motricité fine, comme dans le cas décrit dans l’introduction, que les patients perçoivent comme de la maladresse, des hypesthésies ou des paresthésies à type de fourmillements, jusqu’à un syndrome transversal sensitif ou moteur. Souvent, les poussées durent de quelques jours à plusieurs semaines et régressent spontanément, bien que cette régression puisse rester incomplète. Parallèlement, on observe dans environ 50% des cas une symptomatologie de fatigue dès le début de la maladie, qui entraîne presque toujours une réduction considérable de la qualité de vie des personnes concernées au cours de la maladie (2). Les résultats des recherches actuelles indiquent de plus en plus qu’il existe des parallèles et des recoupements importants dans la physiopathologie.
L’examen clinique révèle souvent une augmentation du niveau des réflexes, des réflexes cutanés abdominaux précoces et un signe de Lhermitte positif (dysesthésie semblable à un choc électrique le long de la colonne vertébrale lors des mouvements de la tête). En cas d’atteinte cérébelleuse, on peut observer un nystagmus, un tremblement d’intention dans l’épreuve doigt-nez ainsi qu’une ataxie de la marche. L'”Expanded Disability Status Scale” (EDSS) sert d’instrument pour l’évaluation standardisée des fonctions cliniques chez les patients atteints de SEP.
Le diagnostic
Selon les critères McDonald actuellement en vigueur, la dissémination temporelle et locale doit être remplie pour établir le diagnostic.
Pour la dissémination locale, l’IRM doit montrer des lésions dans au moins deux des quatre régions typiques de la SEP. En cas de poussée d’une autre localisation, une lésion typique de la SEP suffit. La dissémination temporelle est assurée en présence d’au moins deux lésions d’âges différents (par exemple, présence simultanée de lésions enrichies en produit de contraste et de lésions non enrichies), en présence d’un deuxième événement de poussée (antérieur ou postérieur) ou en cas de détection de bandes oligoclonales spécifiques du liquide (pour plus de détails, voir la publication originale de Thompson et al., 2018) (3).
Dans un premier temps, après une anamnèse détaillée et un examen clinique neurologique, il convient, si la suspicion persiste, de procéder à une IRM avec produit de contraste du crâne ou, selon les symptômes, de la moelle épinière, en posant la question des modifications inflammatoires chroniques. Les quatre localisations classiques des lésions de SEP, généralement ovales, aux limites relativement nettes, puis confluentes, sont juxtacorticales, périventriculaires, infratentorielles et spinales (voir figure 1). Les lésions inflammatoires actives se distinguent par leur prise de contraste (4).
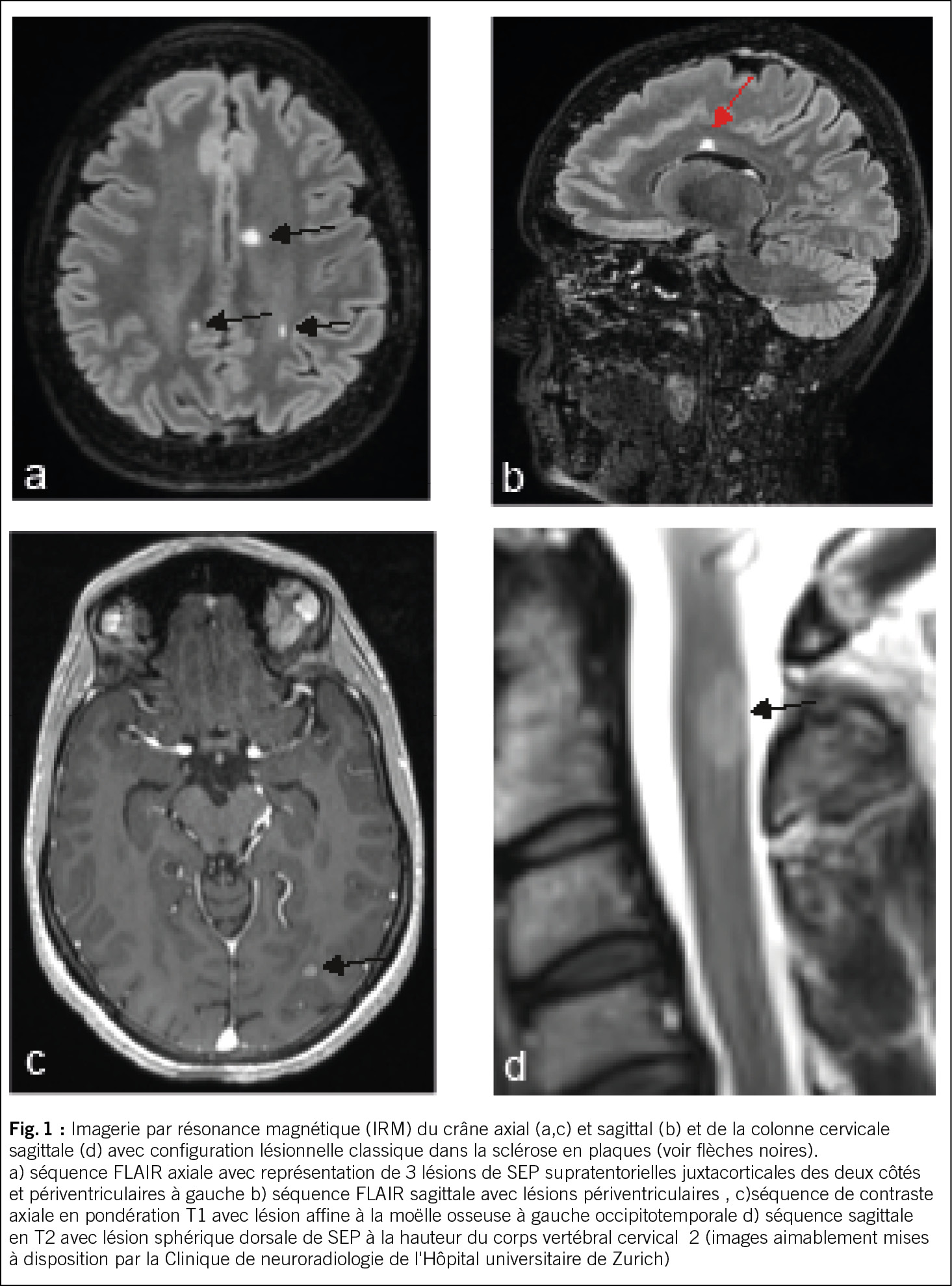
Après avoir été adressé au neurologue, un diagnostic sanguin et un diagnostic du liquide céphalorachidien sont complétés dans le cadre du diagnostic différentiel. Une pléiocytose légère à modérée ainsi qu’une synthèse intrathécale d’immunoglobulines avec mise en évidence de bandes oligoclonales sont typiques. En fin de compte, la SEP est un diagnostic d’exclusion. Les diagnostics différentiels pertinents qui doivent être délimités sont, entre autres, la maladie du spectre de la neuromyélite optique et les maladies associées aux anticorps MOG, les infections du SNC comme la neuroborréliose ou le neurolues et une carence en vitamine B12. Dans de rares cas, des maladies rhumatologiques impliquant le SNC, telles que les vascularites, la sarcoïdose ou la maladie de Behcet, peuvent également provoquer un tableau similaire (5).
Les caractéristiques de base de la thérapie
En cas de survenue d’un épisode de poussée invalidant, un traitement anti-inflammatoire peut être mis en place. On utilise classiquement des stéroïdes à haute dose (par ex. 1g de méthylprednisolone p.o./i.v. pendant 3 à 5 jours). Auparavant, il convient d’exclure une infection et, le cas échéant, une grossesse ; selon le profil de risque, il faut penser à une protection gastrique et à une prophylaxie des thromboses pendant le traitement de la poussée. En cas de régression insuffisante des symptômes, une plasmaphérèse ou une immunoadsorption peuvent être évaluées en tant que procédure de réserve.
Pour prévenir les poussées, de nombreuses préparations modifiant l’évolution de la maladie sont désormais disponibles et permettent d’influencer considérablement le cours de la maladie. Il est désormais clair qu’un début de traitement précoce a un effet plus favorable sur l’évolution de la maladie qu’un début de traitement retardé (“hit hard and early”) (6)(7).
Les médicaments autorisés sont classés en trois catégories par ordre croissant d’efficacité. Le choix du traitement se fait d’une part en fonction de l’activité présumée de la maladie sur la base de la clinique et de l’imagerie, et d’autre part en fonction de facteurs personnels (en particulier le désir d’enfant ou la grossesse). Outre les préparations à base d’interféron établies depuis les années 1990, la catégorie 1 comprend notamment le fumarate de diméthyle et le tériflunomide. Les modulateurs du récepteur de la sphingosine-1-phosphate les plus récents (par ex. le fingolimod) sont attribués à la catégorie 2. Les substances de la catégorie 3 sont le natalizumab, un inhibiteur de l’intégrine alpha4 qui inhibe la migration des lymphocytes T à travers la barrière hémato-encéphalique, et les anticorps monoclonaux anti-CD20, qui provoquent une déplétion des cellules B.
Le pronostic des personnes atteintes s’est nettement amélioré au cours des dernières décennies grâce à la disponibilité des préparations modulant la maladie, de sorte que l’on peut aujourd’hui s’attendre à une espérance de vie non compromise au moment du diagnostic. Un début précoce et monosymptomatique de la maladie avec une régression complète des symptômes de poussée s’est avéré être un facteur pronostique favorable (7). En complément, des mesures thérapeutiques non médicamenteuses, par exemple la physiothérapie et l’ergothérapie, ainsi que des stratégies thérapeutiques de soutien pour le contrôle des symptômes sont essentiels et nécessitent une prise en charge interdisciplinaire étroite.
L’ auteur a publié cet article en allemand dans « der informierte arzt – die informierte ärztin» 12_2023, la traduction en français a été réalisée par les éditeurs. L’ auteur n’ assume aucune responsabilité pour les modifications dues à une traduction.
Copyright Aerzteverlag medinfo AG
Médecin-chef, Clinique de neurologie
Hôpital universitaire de Zurich
Frauenklinikstrasse 26
8091 Zurich
Médecin-chef, Clinique de neurologie
Hôpital universitaire de Zurich
Frauenklinikstrasse 26
8091 Zurich
Les auteurs n’ont pas déclaré de conflits d’intérêts en rapport avec cet article.
1. Schweizerische Multiple Sklerose Gesellschaft [Internet]. 2023 [cited 2023 Oct 26]. Available from: https://www.multiplesklerose.ch/de/
2. McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and Treatment of Multip¬le Sclerosis: A Review. JAMA. 2021 Feb 23;325(8):765–79.
3. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018 Feb;17(2):162–73.
4. Filippi M, Rocca MA, Ciccarelli O, De Stefano N, Evangelou N, Kappos L, et al. MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet Neurol. 2016 Mar;15(3):292–303.
5. DGN One | Leitlinie Details [Internet]. [cited 2023 Oct 26]. Available from: htrum-erkrankungen-und-mog-igg-assoziierten-erkrankungen
6. Amato MP, Fonderico M, Portaccio E, Pastò L, Razzolini L, Prestipino E, et al. Di¬sease-modifying drugs can reduce disability progression in relapsing multiple scle¬rosis. Brain J Neurol. 2020 Oct 1;143(10):3013–24.
7. Wiendl H, Gold R, Berger T, Derfuss T, Linker R, Mäurer M, et al. [Multiple sclerosis treatment consensus group (MSTCG): position paper on disease-modifying treatment of multiple sclerosis 2021 (white paper)]. Nervenarzt. 2021 Aug;92(8):773–801.


la gazette médicale
- Vol. 13
- Ausgabe 2
- April 2024