Das Mayer-Rokitansky-Küster-Hauser-Syndrom (MRKH) ist eine einschneidende Diagnose für Jugendliche mit weitreichenden Konsequenzen. Das MRKH-Syndrom wird meist entdeckt durch das Ausbleiben der Menstruation in der Pubertät und wird in zwei Formen eingeteilt. Als Therapie gibt es die konservativen Dehnungsbehandlungen sowie chirurgisch-minimal invasive Techniken, welche unabhängig des operativen Ansatzes einer intensiven Nachsorge bedürfen.
Mayer-Rokitansky-Küster-Hauser syndrome (MRKH) is a drastic diagnosis for young people with far-reaching consequences. MRKH syndrome is usually discovered due to the absence of menstruation during puberty and is categorised into two forms. There are conservative stretching treatments as well as surgically minimally invasive techniques, which require intensive aftercare regardless of the surgical approach.
Key words: Mayer-Rokitansky-Küster-Hauser-Syndrom, Vecchietti Operation, Shears Operation, Dilatation nach Frank, Amenorrhoe
Krankheitsbild
Das MRKH-Syndrom ist eine Anomalie der Müller’schen Gänge. Diese zeichnet sich durch eine angeborene Aplasie des Uterus und der oberen 2/3 der Vagina aus bei einer phänotypisch weiblichen Person mit den Geschlechtschromosomen XX (1). Diese Malformation wurde bereits im 11. Jahrhundert von Albucassis und Avicenna beschrieben, jedoch ist das vollständige MRKH-Syndrom erst seit etwa 150 Jahren bekannt. Der Name wurde 1961 nach der Präzisierung der Definition durch Hauser und Schreiner von «uterus bipartitus solidus rudimentarius cum vagina solida» in Mayer-Rokitansky-Küster-Hauser-Syndrom geändert, benannt nach den Erstbeschreibern (2). Das MRKH-Syndrom hat eine Inzidenz von 1:5’000, welche in zwei gross angelegten, europäischen epidemiologischen Studien erhoben wurde (3, 4).
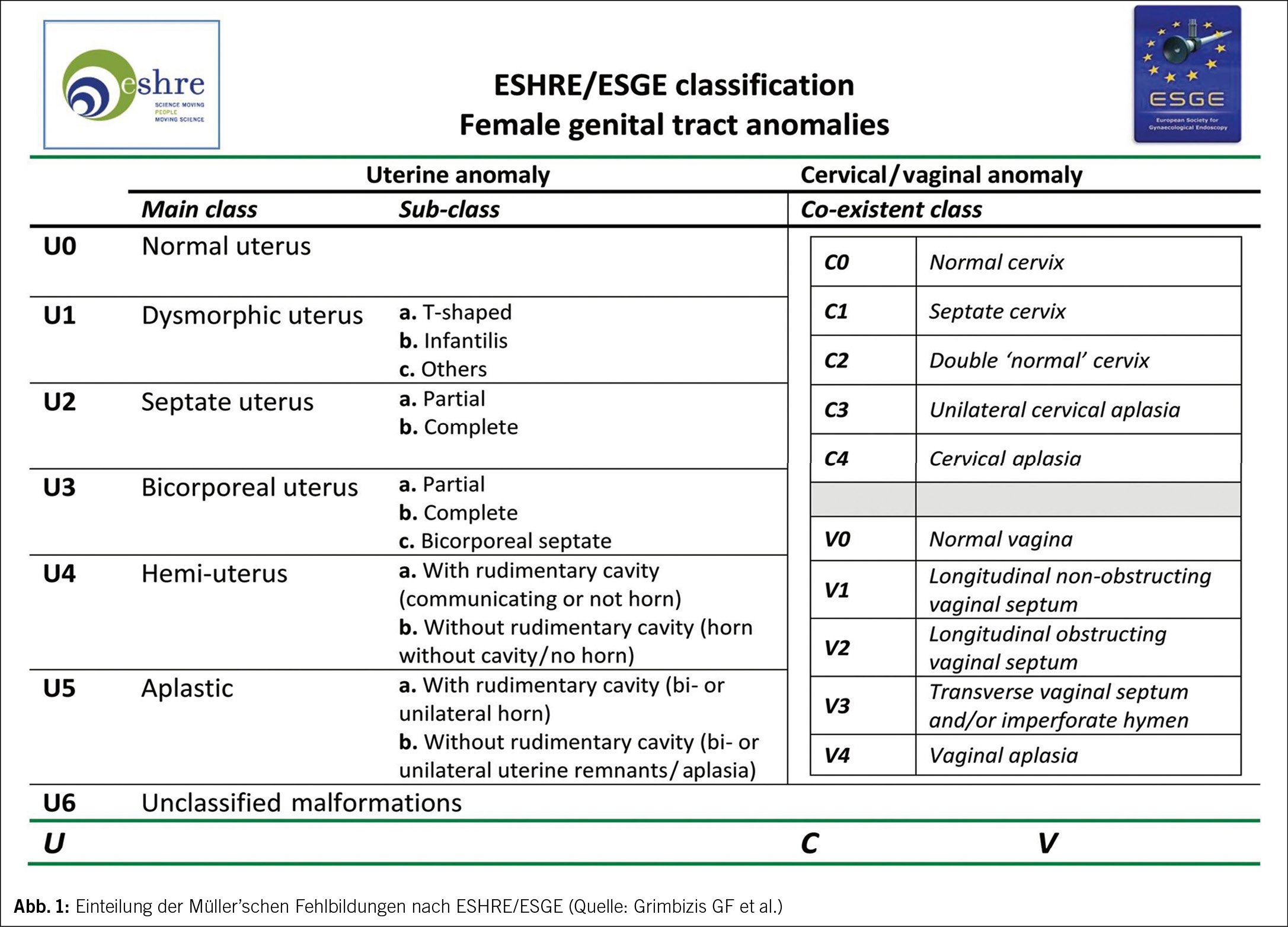
Gemäss der ESHRE/ESGE-Klassifikation von 2013 wird die Anomalie als U5 C4 V4 klassifiziert (5). Die ESHRE/ESGE-Guidelines lassen alle Kombinationen von Müller’schen Anomalien klassifizieren (Abb. 1).
Embryologie und Genetik
In der Embryologie bildet sich der paramesonephrische Duct (Müller’scher Gang) ab der 5. / 6. Schwangerschaftswoche aus. Familiäre Konstellationen deuten auf eine monogenetische Ursache hin. Im Weiteren gibt es aber auch Hinweise auf polygenetische und multifaktorielle Faktoren als auch Umgebungsfaktoren sowie regulatorische Mechanismen, wo epigenetische und somatische Ereignisse eine Rolle spielen (6). In Tiermodellen wurden verschiedene Faktoren nachgewiesen. Die sich ständig entwickelnden Möglichkeiten genetischer Abklärung sind mit den Betroffenen zu besprechen und dürften das Wissen um mögliche Faktoren in Zukunft erweitern. Dies dürfte auch von Bedeutung sein, falls die Uterus-Transplantation in Zukunft für Betroffene zur Verfügung stehen wird.
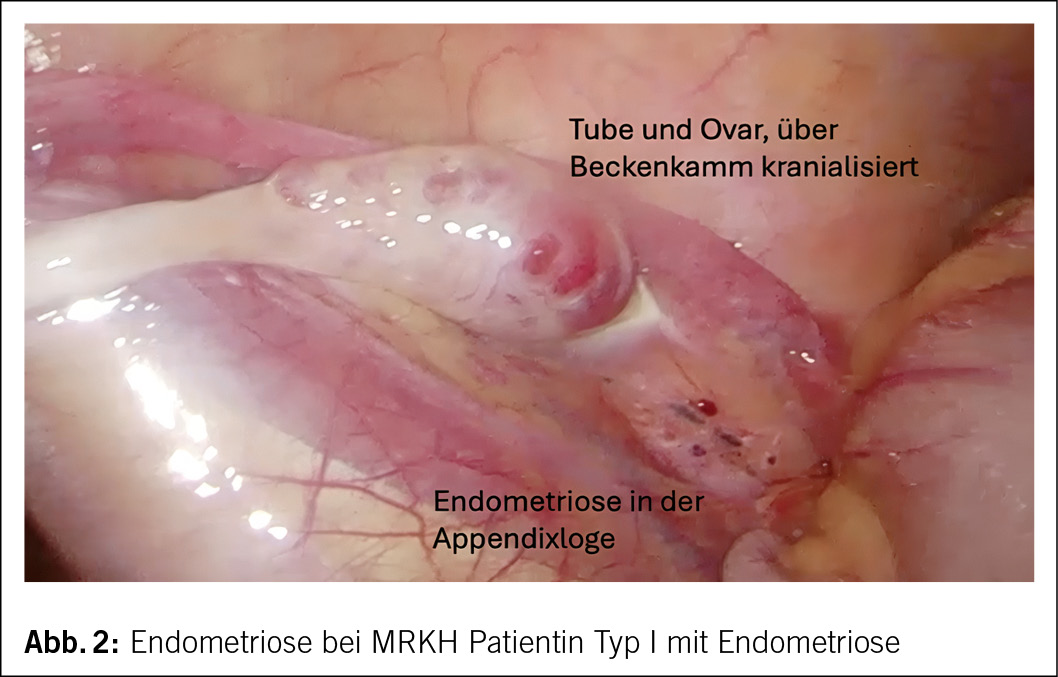
Beim MRKHS werden zwei Formen unterschieden: Typ I, das isolierte Fehlen von Vagina und Uterus (ca. 60 % der Betroffenen) und Typ II, das atypische MRKHS mit asymmetrischer Hypoplasie einer oder beider Uterusknospen sowie Tubendysplasie zusätzlich zur Vaginalaplasie (ca. 40 % der Betroffenen). Diese Form ist mit weiteren Fehlbildungen verbunden, insbesondere im oberen Harntrakt und Skelettsystem. Eine schwerwiegende Form ist die MURCS-Assoziation (Müllerian-Renal-Cervicalthoracic-Somite abnormalities), bei der auch Herzanomalien und Hörverlust auftreten können.
Diagnostisches Work-up und Transition
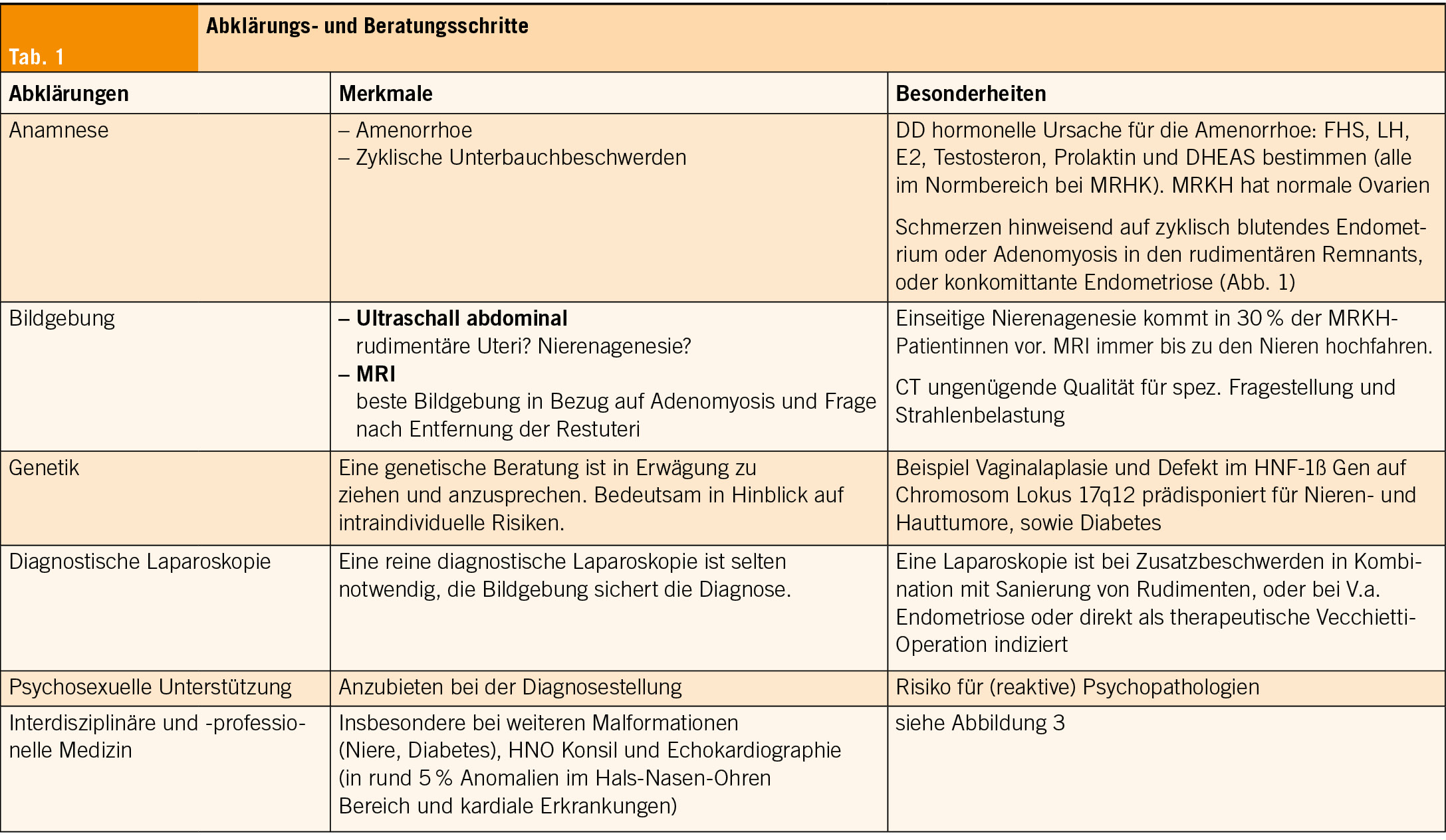
Die meisten MRKH-Diagnosen werden zwischen dem Alter von 15 und 17 Jahren gestellt, wenn der Übergang von der Kinder- in die Erwachsenenmedizin stattfindet. Die diagnostischen Schritte können von jugend-gynäkologisch geschulten PädiaterInnen oder GynäkologInnen eingeleitet werden (Tabelle 1). Nach einer wegweisenden Anamnese folgen Blutuntersuchungen zum Hormonstatus und die Bildgebung. In der Bildgebung hat sich das MRI etabliert, da es mögliche Zusatzanomalien oder kranialisierte Ovarien, welche bei fehlender Vagina sonographisch nicht einfach erhoben werden können, visualisieren lässt (7). Differentialdiagnostisch ist an XY DSD (disorder of sex differenciation) zu denken. Es handelt sich dabei um eine seltene angeborene Genitalfehlbildung beim männlichen Geschlecht. Eine ebenfalls XY-assoziierte Ausprägung kann das CAI-Syndrom sein (CAIS = complete androgen insensitivity syndrome, auch Morris Syndrome genannt), wo eine Mutation im Androgenrezeptor Gen vorliegt. Phänotypisch sind die Personen weiblich mit blind endender kurzer Vagina und wenig Schambehaarung.
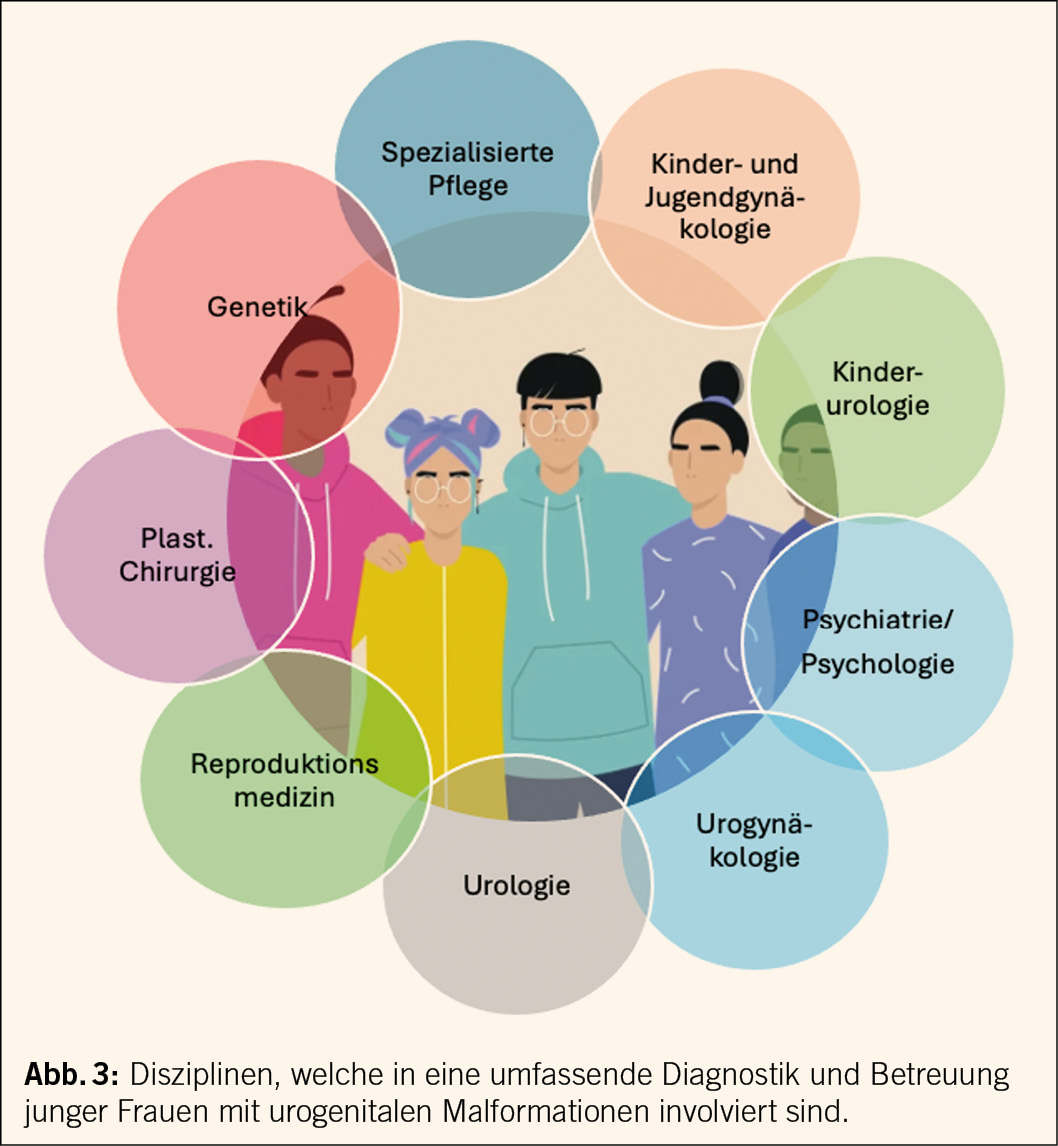
Die Diagnose MRKH trifft die jungen Frauen in der Adoleszenz, einer vulnerablen Lebensphase. Die Diagnose kann Betroffene in eine ernsthafte Krise stürzen, sind doch die Identität, die Sexualität und die Reproduktion betroffen. Eine sensible psychologische Begleitung durch eine hierzu geschulte Person ist anzusprechen und zu empfehlen. Es liegen bei MRKH-Betroffenen häufiger Ängste, Depression und Minderwertigkeitsprobleme vor (8).
Therapie des MRKH
Der optimale Zeitpunkt für die Durchführung einer Neovaginoplastik liegt nach Abschluss der Pubertätsentwicklung vor, sofern eine ausreichende Östrogenproduktion gewährleistet ist und die Patientin den Wunsch nach sexuellen Aktivitäten äussert. Die erfolgreiche Umsetzung dieser Therapie erfordert die Einwilligungsfähigkeit und das Verständnis der Patientin, bei den Therapien einen aktiven Part einzunehmen, speziell was die Nachbehandlung mit Dilatationen betrifft. Auf diesen wichtigen Teil der Behandlung wird untenstehend weiter eingegangen. Die OP soll auf expliziten Wunsch der Betroffenen durchgeführt werden, denn eine vorzeitige Operation auf Wunsch der Eltern erhöht das Risiko von Misserfolgen, traumatischen Erfahrungen für die Patientin sowie die Notwendigkeit wiederholter Eingriffe zur Aufrechterhaltung der Vaginalintegrität, welche für ein späteres zufriedenstellendes Sexualleben ein entscheidender Faktor ist. Unabhängig davon, ob konservative oder operative Verfahren angewendet werden, erfordert die Durchführung der Vaginaldilatation im Verlauf beträchtliche Anstrengungen, insbesondere bei jüngeren Frauen, und eine mal-compliance kann den Therapieerfolg erheblich schmälern. Ziel ist eine Vaginallänge von mindestens 6.6 cm zu bekommen, da ab dieser Länge die sexuelle Zufriedenheit hoch war (9).
Nicht-invasive Therapie
Bei Patientinnen mit präsentem Scheidengrübchen und einem zentral gelegenen Meatus urethrae externus kann eine Dilatationstherapie angeboten werden. Die erste Beschreibung eines solchen Verfahrens erfolgte 1938 durch Frank und wurde dann durch Ingram weiterentwickelt. Die Therapie dauert 4–6 Monate, bis eine suffiziente Vaginallänge erreicht wird, welche penetrative Kohabitationen ermöglicht.
Täglich sollte für mindestens 30 Minuten ein Druck bzw. eine Dilatation des Vaginalgrübchens erfolgen. Hierzu sollte das Vaginalgrübchen nicht vernarbt sein. Dieses lange dauernde Verfahren hat Erfolgsquoten von bis zu 95 % und aufgrund der nicht-invasiven Natur der Methode wird sie z.B. in den USA als Methode der ersten Wahl angesehen (10). Es muss aber beachtet werden, dass die Therapie in ungefähr 15 % nicht erfolgreich ist, resp. abgebrochen wird.
In Studien zur sexuellen Zufriedenheit zeigte die Dilatationsmethode zwar die niedrigsten Werten im FSFI-Score (25.7 Punkte) bei gleichzeitig kürzester mittlerer Vaginallänge von 6.7 cm (11, 12). Der Female Sexual Function Index (FSFI) misst die sexuelle Zufriedenheit anhand eines Scores mit Werten zwischen 0–36. Je höher die Punktzahl, umso grösser ist die sexuelle Zufriedenheit. Dabei ist zu erwähnen, dass bei den in diesem Artikel diskutierten Methoden die FSFI-Werte in Studien ähnlich waren, nämlich zwischen 25.7 und 29.9 Punkten. Bei der Dilatationsmethode wird die Neovagina mit normalem Vaginalepithel ausgekleidet und bietet ein normales vaginales Milieu.
Operative Techniken
Das Verfahren nach Wharton-Shears-George basiert auf dem schrittweisen Dilatieren der rudimentär vorliegenden Müller-Gänge mit Hegarstiften bis auf Grösse 14 in jedem Kanal. Dann wird durch Inzision zwischen beiden Hegarstiften und Präparation in die Tiefe eine Neovagina im Spatium vesikorektale kreiert. Begrenzende Strukturen sind die hintere Blasenwand und die vordere Rektumwand, welche auf Integrität am Ende des Eingriffs kontrolliert werden müssen. Ein perinealer Hautlappen kann in die Neovagina transplantiert werden, um die Epithelialisierung zu verbessern. Ein Vaginalstent beschichtet mit Östrogencrème wird für ungefähr eine Woche eingelegt und danach durch grössere Dilatatoren ersetzt.
Im Verlauf wird empfohlen täglich zu dilatieren, da das Gewebe zu schrumpfen neigt. Dies auf alle Fälle, bis regelmässige Kohabitationen aufgenommen werden. Die Neovagina wird innerhalb von mehreren Monaten vom Introitusbereich aus mit physiologischem nicht-verhorntem Vaginalepithel epithelialisiert und erreicht im Mittel eine Länge von 8.3 cm (13). Diese Methode hatte in Studien einen hohen FSFI-Score von 29.9 Punkten bei 289 Frauen.
Eine Kombination aus operativer Anlage einer Neovagina mit einem Dehnungsverfahren bietet die Methode nach Vecchietti. Hierbei wird ein Phantom in die Vaginalgrube eingelegt und daran angebrachte Fäden auf Höhe des Bauchnabels mittels Laparoskopie ausgeführt. Mit Hilfe einer Spannungsapparatur wird jeden Tag der Zug auf die Fäden erhöht und somit eine Vaginaldehnung von bis zu 1 cm/Tag erreicht. Ziel ist, eine Vaginallänge von 10-12 cm zu erhalten. Die Phase der raschen Vaginaldehnung erfolgt unter stationären Bedingungen bei liegender Epiduralanästhesie (EDA) zur Vermeidung von Schmerzen. Heutzutage werden walking-EDAs angelegt, um eine längere Immobilität zu vermeiden. Wie bei der Wharton-Shears-George-Methode erfolgt die Epithelisierung vom Introitus aus mit normalem Vaginalepithel.
Es wird nach Entfernung des Spannapparates in den ersten 3 postoperativen Monaten empfohlen, 24 Stunden pro Tag mit reichlich östrogenhaltiger Creme ein Phantom zu tragen. Sollte nach der Abheilungs-/Epithelialisierungsphase keine regelmäßige Kohabitation möglich sein, sollte das Phantom zumindest nachts für einige weitere Monate getragen werden, da sonst ein Risiko für eine sekundäre Schrumpfung der Neovagina besteht. Die Länge und Breite des Dobbies lassen wir am Universitätsspital Zürich nach Entfernung des Spannapparates individuell anpassen.
Auch zeigten Studien zur sexuellen Zufriedenheit normale Werte im FSFI-Score von 29.8, bei einer mittleren Vaginallänge 7.9 cm. Urologische Komplikationen bei der Anlage des Spannapparates traten in 2 % der Fälle auf (11).
Bei der Darmscheidenanlage wird ein Darmabschnitt (in der Regel Sigma oder Ileum) isoliert und für die Konstruktion eines Vaginalkanals verwendet. Die Darmscheidenanlage ist unter den bisher vorgestellten Methoden die aufwändigste Operation. Diese Technik ist besonders in der Kinderchirurgie eine etablierte Methode. Das Darmsegment wird sorgfältig vorbereitet, um die Blutversorgung und die Integrität der Schleimhaut sicherzustellen. Anschliessend wird es chirurgisch in die Beckenhöhle implantiert, um die Neovagina zu bilden. Im Laufe der Zeit heilt die Neovagina und passt sich der Funktion eines natürlichen Vaginalkanals an, wobei Studien die längste Vaginallängen zeigten im Vergleich zu den anderen beschriebenen Techniken von 12.9 cm mit einem FSFI-Score von 27.8 (12). Zu beachten ist, dass diese Technik die höchsten Dyspareunieraten aufweist und Stenosen in knapp 10 % der Fälle im Verlauf vorkommen. Vor geplanter Uterus-Transplantation sollten diese Verfahren nicht angewandt werden.
Komplikationen oder unerwünschte Begleiterscheinungen reichen von mukösem, teils übelriechendem Ausfluss bis hin zu Strikturen oder Nekrosen im Anastomosenbereich. Darmkrebsfrüherkennungsuntersuchungen sind nach dieser Methode empfohlen.
Copyright Aerzteverlag medinfo AG
Oberarzt Klinik für Gynäkologie
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
Stellvertretende Klinikdirektorin
Klinik für Gynäkologie, USZ
Frauenklinikstrasse 10
8006 Zürich
cornelia.betschart@usz.ch
Die Autoren haben keine Interessenkonflikte angegeben.
1. Herlin MK, Petersen MB, Brannstrom M. Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: a comprehensive update. Orphanet journal of rare diseases. 2020;15(1):214.
2. Hauser GA, Schreiner WE. (Mayer-Rokitansky-Kuester syndrome. Rudimentary solid bipartite uterus with solid vagina). Schweiz Med Wochenschr. 1961;91:381-4.
3. Herlin M, Bjorn AM, Rasmussen M, Trolle B, Petersen MB. Prevalence and patient characteristics of Mayer-Rokitansky-Kuster-Hauser syndrome: a nationwide registry-based study. Hum Reprod. 2016;31(10):2384-90.
4. Aittomaki K, Eroila H, Kajanoja P. A population-based study of the incidence of Mullerian aplasia in Finland. Fertil Steril. 2001;76(3):624-5.
5. Grimbizis GF, Gordts S, Di Spiezio Sardo A, Brucker S, De Angelis C, Gergolet M, et al. The ESHRE/ESGE consensus on the classification of female genital tract congenital anomalies. Hum Reprod. 2013;28(8):2032-44.
6. Cunha GR, Robboy SJ, Kurita T, Isaacson D, Shen J, Cao M, et al. Development of the human female reproductive tract. Differentiation. 2018;103:46−65.
7. Preibsch H, Rall K, Wietek BM, Brucker SY, Staebler A, Claussen CD, et al. Clinical value of magnetic resonance imaging in patients with Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: diagnosis of associated malformations, uterine rudiments and intrauterine endometrium. European radiology. 2014;24(7):1621-7.
8. Heller-Boersma JG, Schmidt UH, Edmonds DK. Psychological distress in women with uterovaginal agenesis (Mayer-Rokitansky-Kuster-Hauser Syndrome, MRKH). Psychosomatics. 2009;50(3):277-81.
9. Callens N, De Cuypere G, Wolffenbuttel KP, Beerendonk CC, van der Zwan YG, van den Berg M, et al. Long-term psychosexual and anatomical outcome after vaginal dilation or vaginoplasty: a comparative study. The journal of sexual medicine. 2012;9(7):1842-51.
10. Edmonds DK, Rose GL, Lipton MG, Quek J. Mayer-Rokitansky-Kuster-Hauser syndrome: a review of 245 consecutive cases managed by a multidisciplinary approach with vaginal dilators. Fertil Steril. 2012;97(3):686-90.
11. McQuillan SK, Grover SR. Dilation and surgical management in vaginal agenesis: a systematic review. International urogynecology journal. 2014;25(3):299-311.
12. McQuillan SK, Grover SR. Systematic review of sexual function and satisfaction following the management of vaginal agenesis. International urogynecology journal. 2014;25(10):1313-20.
13. Walch K, Kowarik E, Leithner K, Schatz T, Dorfler D, Wenzl R. Functional and anatomic results after creation of a neovagina according to Wharton-Sheares-George in patients with Mayer-Rokitansky-Kuster-Hauser syndrome-long-term follow-up. Fertil Steril. 2011;96(2):492-7 e1.


info@gynäkologie
- Vol. 14
- Ausgabe 3
- Juni 2024