

Die Glasknochenkrankheit oder Osteogenesis imperfecta (OI) ist eine heterogene Gruppe von klinischen Manifestationen verschiedener genetischer Veränderungen. Im Vordergrund stehen erhöhte Brüchigkeit und Deformierbarkeit von Knochen, aber auch Wachstumsstörungen und extraossäre Veränderungen. Ursächlich liegt bei 90% der Fälle eine autosomal-dominante Mutation in den für die Bildung von Kollagenfibrillen massgebenden Genen COL1A1 / COL1A2 vor. Bisphosphonate sind derzeit die beste medikamentöse Therapieoption für schwerere Formen von OI. Im diesem Artikel kommen die für die Erfassung und Behandlung wesentlichsten Punkte zur Darstellung.
Unter der Diagnose einer Osteogenesis imperfecta wird eine heterogene Gruppe von Phänotypen, verursacht durch Mutationen /Sequenzvarianten in verschiedenen Genen subsumiert, welche sich primär am Skelett manifestieren, aber letztlich generalisierte Bindegewebserkrankung sind. Im Vordergrund steht eine quantitativ und qualitativ verminderte Knochenmasse mit Knochenbrüchigkeit, welche sich durch Neigung zu Frakturen der langen Röhrenknochen und Kompressionsfrakturen der Wirbelkörper ohne adäquate Traumata manifestiert. Dazu gesellen sich Deformierbarkeit der langen Knochen, Rippen und Wirbelsäule sowie Kleinwuchs. Sekundäre Merkmale können Muskelschwäche, überstreckbare Gelenke, dünne Haut mit vermehrter Gefässbrüchigkeit und Blutungsneigung, bläuliche Skleren, Schallleitungs- oder gemischte Schwerhörigkeit, gestörte Zahnbildung mit durchschimmernden Zähnen und Malocclusion, Skoliose, Lungenfunktionsstörungen und Abnormitäten der Herzklappen sein.
Epidemiologie und Pathogenese
Etwa 1 von 10 000 Neugeborenen weist eine mit OI assoziierte genetische Konstellation auf (1–3), was in der Schweiz zu 300–400 Betroffenen führt. Diese Zahl kann in abgeschlossenen Regionen mit einem erhöhten Grad an Blutverwandtschaft höher liegen. Schwere Formen können bereits in der Perinatalperiode zum Tod führen oder präsentieren sich im Kleinkindesalter mit multiplen Frakturen ohne adäquates Trauma. Milde Formen können sich erst im frühen Erwachsenenalter mit einer Osteoporose manifestieren (1, 2, 4).
Kollagen Typ I ist ein wichtiges Strukturprotein für Knochen, Sehnen, Ligamente, Haut und Skleren. Kollagenfasern von Typ I sind Polymere von Tropokollagen-Molekülen, welche aus je zwei Alpha-1- und einer Alpha-2-Kette in Trippelhelixform gebildet sind. Den weitaus meisten Fällen von OI liegen autosomal dominante (AD) Defekte der diese Alpha-Ketten codierenden Gene COL1A1 und seltener COL1A2 zugrunde. Sie beeinflussen die Quantität oder Struktur der entsprechenden Typen von Alphaketten und damit direkt der Kollagenfasern Typ I. Nur bei rund 10% der OI sind die Gene COL1A1 und COL1A2 normal (2, 4–6).
Diese kleinere Gruppe von OI basieren auf einer Vielzahl von meistens rezessiven Gendefekten und betreffen einerseits die zelluläre «Maschinerie» der posttranslationalen Bearbeitung der Bestandteile der Kollagenfasern mit Defekten in Aufbau, Reifung, Transport und Sekretion, andererseits aber selten andere an der Bildung und Homöostase von Knochen und Knorpel beteiligte Proteine (1, 2, 4).
Art und Ausmass der Manifestationen von Gendefekten sind sowohl vom exakten Genlocus wie auch von der Art der Mutation abhängig, was die enorme Breite des Spektrums klinischer Manifestationen zu einem grossen Teil erklären kann. Darüber hinaus ist in diesem Zusammenhang die Tatsache bemerkenswert, dass sich die klinischen Manifestationen sogar bei identischen Gendefekten stark unterscheiden können, was darauf hindeutet, dass nicht isolierte Mutationen alleine, sondern vielmehr das Zusammenspiel einer Vielzahl von an der Bildung der Weichteile beteiligten Komponenten von ausschlaggebender Bedeutung ist (2, 4).
Klassifikation, klinische Manifestation und Diagnose
Historisch standen klinische Klassifikationssysteme im Vordergrund, in den 70er Jahren wurden von Sillence 4 OI-Typen unterschieden (7), welche durch die klinische Symptomatik, radiologische Befunde und Vererbungsmuster charakterisiert waren (Tabelle 1). Die Einteilung nach Sillence wurde im Laufe der Jahre erweitert und mit den Fortschritten der Genetik wurden auch Vorschläge für genetische Klassifikationssysteme publiziert (Tabelle 2) (2, 8). Da im klinischen Alltag die Beurteilung und die therapeutischen Konsequenzen des individuellen Patienten jedoch weiterhin primär auf klinischen Befunden basieren, wird aktuell von vielen Autoren eine Einteilung nach genetisch-funktionellen Gesichtspunkten bevorzugt (6, 8, 9, 38, 39). Dabei repräsentieren die Typen I-IV Fälle mit autosomal-dominanten Mutationen von COL1A1/2 und andere und noch neu zu entdeckende Gendefekte werden weiteren Typennummern zugeordnet. Unter dem Typ I wird eine milde, dem Typ II eine perinatal letale, dem Typ III eine schwere und dem Typ IV eine moderate Verlaufsform subsumiert.
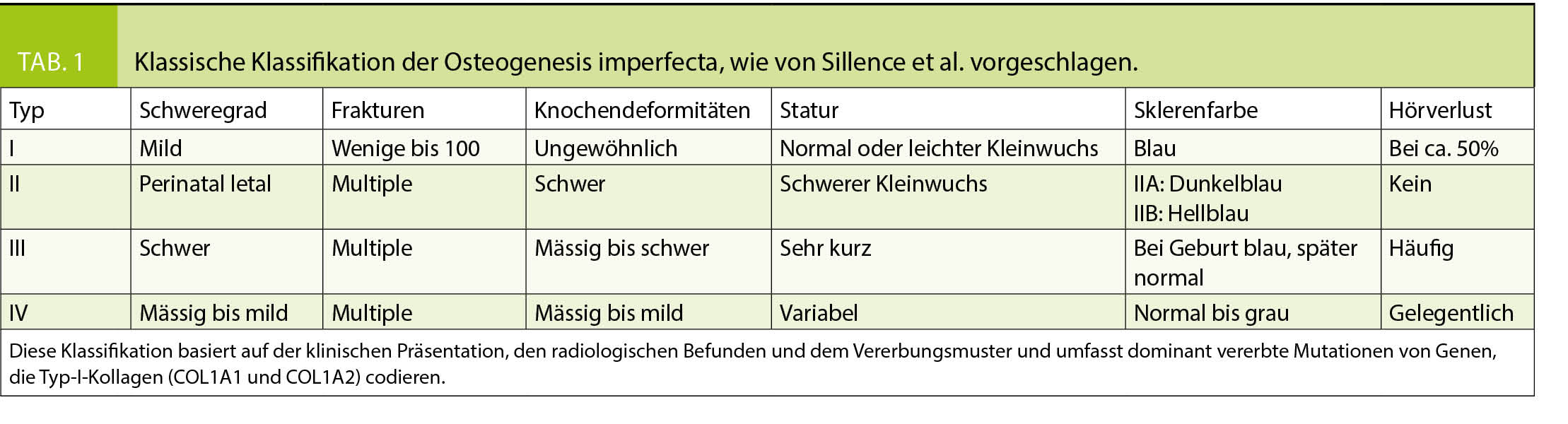
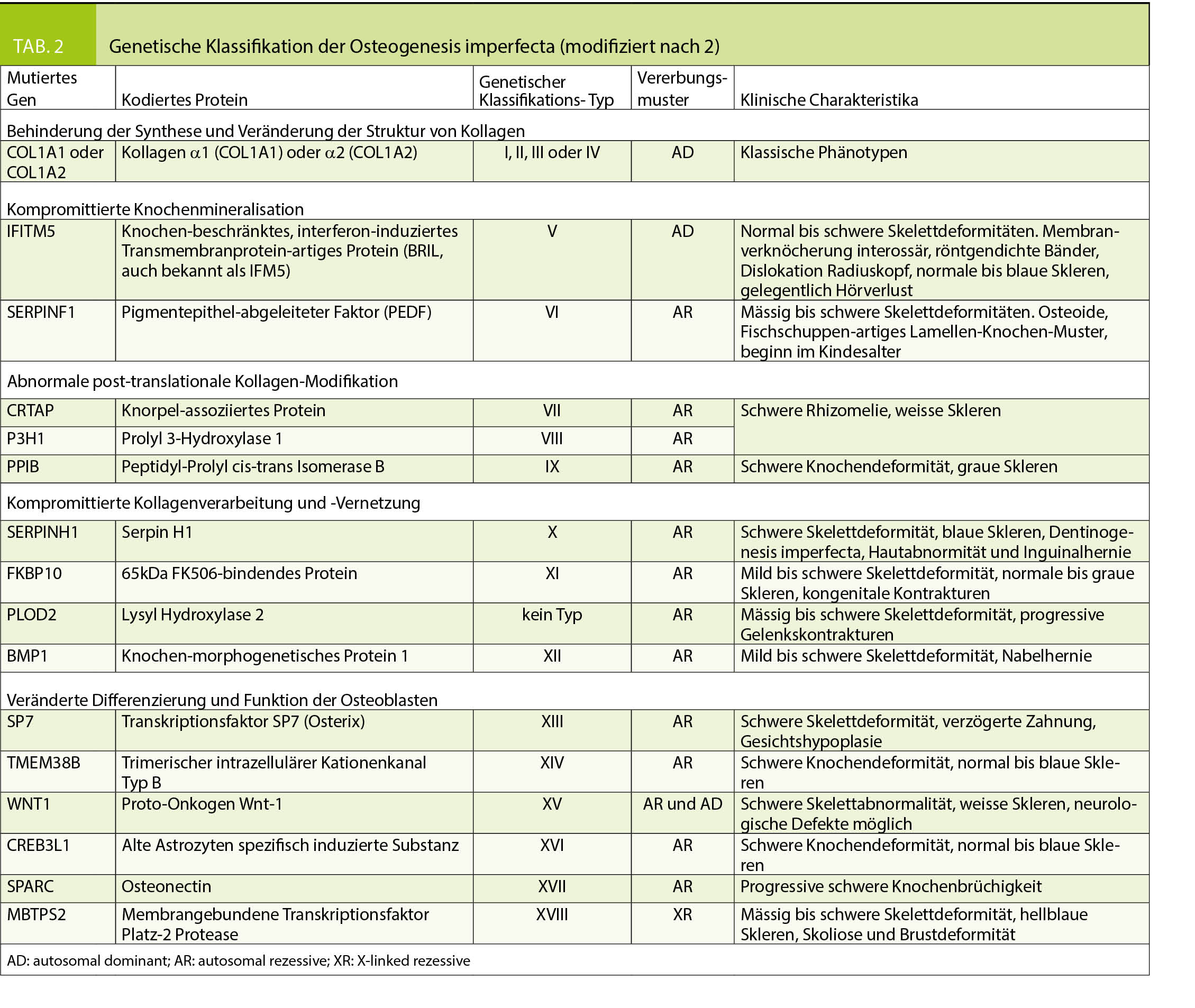
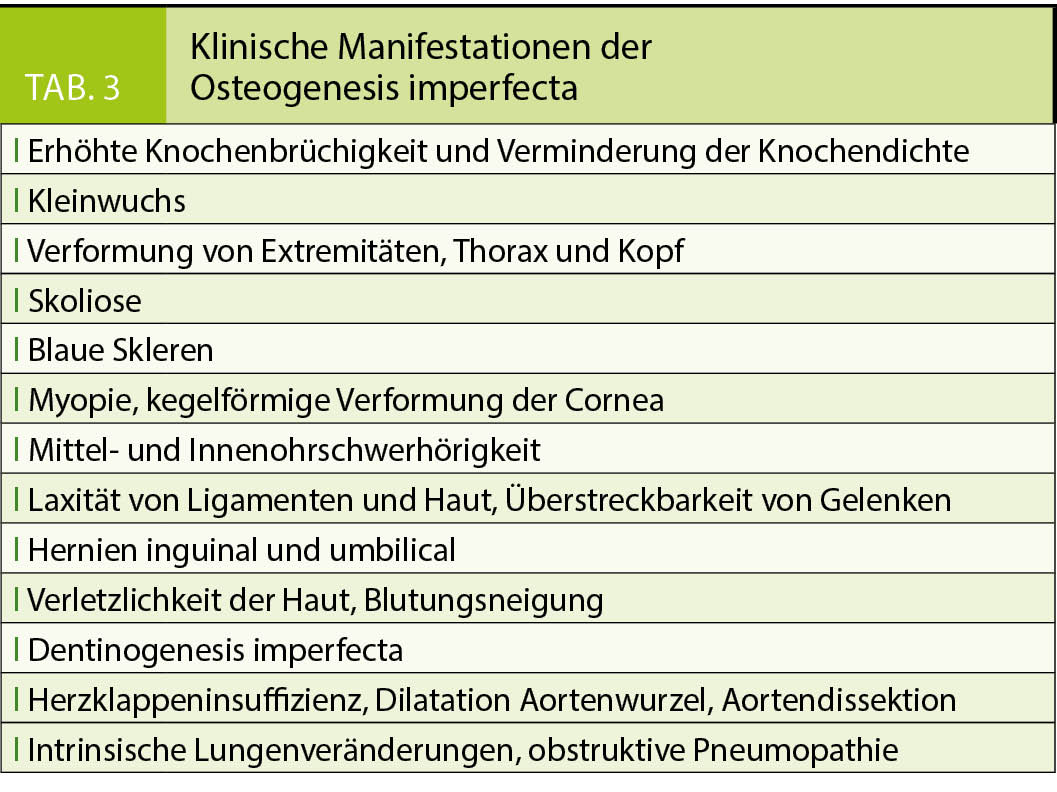
Tabelle 3 fasst die wesentlichen klinischen Manifestationen der OI zusammen (1, 8–11). Während sich die Typen II und höher regelhaft schon im (Klein)Kindesalter manifestieren, können Genträger des Typs I zwar bereits intrauterin und beim ersten Gehen Frakturen erleiden, jedoch das Erwachsenenalter auch unerkannt erreichen. Deformitäten sind geringgradig, die Statur meistens normal. Bei oligosymptomatischen Erwachsenen kann sich die Krankheit postmenopausal in Form einer beschleunigten Osteoporose manifestieren. Auch vorzeitige Hörstörungen ab der zweiten bis vierten Dekade sind typisch. Weitere Kennzeichen können rasche Alterung, Arthrosen und Bandlaxität sein. Bei dieser Form kann es im Verlauf zu einer erhöhten Frakturinzidenz und einem erheblich erniedrigten Knochenmineralgehalt kommen, welcher nach Ausschluss anderer Ursachen diagnostisch in Richtung OI wegweisend sein kann. Dabei kann die Bestimmung mittels DEXA durch Knochendeformitäten, Skoliose, Kleinwuchs, vorbestehende Frakturen oder Osteosynthesematerial erschwert sein.
Die klinische Diagnose basiert auf den erwähnten Symptomen und Zeichen. Bei Individuen mit multiplen Frakturen, einer positiven Familienanamnese und typischen extraossären Manifestationen ist die Diagnose leicht zu stellen, bei Fehlen derselben und geringen und unspezifischen extraossären Zeichen schwierig (1, 8–11). Insbesondere bei älteren Jugendlichen und jungen Erwachsenen mit einer vorzeitigen, gelegentlich als idiopathisch bezeichneten Osteoporose ist an die Möglichkeit einer OI v. a. des Typs I zu denken. Ein die OI beweisender Labortest ist nicht leicht zugänglich. Wohl können verschiedene Parameter des Knochen- und Mineralstoffwechsels und Marker von Knochenbildung und -abbau pathologisch ausfallen, aber nicht in einem Muster mit adäquater Sensitivität und Spezifität. Pyridinoline im Urin werden als mögliche Biomarker für OI diskutiert (41). Heute werden in Speziallabors molekulargenetische Analysen auf Basis einer «Next-Generation»-Sequenzierung aller bekannter, mit einer OI assoziierten Gene als Gen-Panels angeboten, welche eine rasche Diagnose erlauben (1, 2, 12, 13). Bedingt durch eine Limitatio können Gen-Panel Analysen nur durch Ärzte mit eidgenössischem Weiterbildungstitel «Medizinische Genetik» verordnet werden. Vorgängig sollen Ursachen einer möglichen sekundären Osteoporose immer ausgeschlossen werden und muss ein dringender Verdacht auf eine OI bestehen. Falls dabei Mutationen mit unklarer Funktion entdeckt werden, stehen biochemische Untersuchungen zum Nachweis von Veränderungen der Menge, Struktur und von posttranslationalen Modifikationen von Prokollagen I zur Verfügung, das Ausgangsmaterial wird aus Kulturen von bioptisch entnommenen Fibroblasten gewonnen.
Die Differentialdiagnose umfasst eine Reihe von Zuständen, welche ebenfalls mit einer fragilen Knochenstruktur einhergehen. Erwähnt seien beispielhaft die Rachitis, die Osteomalazie, ein juveniler M. Paget, die seltene AD-vererbte Hypophosphatasie und die idiopathische juvenile Osteoporose. Bei Kindern mit multiplen Frakturen in verschiedenen Heilungsstadien, wie sie bei OI gesehen werden, muss auch an die Möglichkeit einer Kindsmisshandlung gedacht werden und umgekehrt bei Verdacht auf Kindsmisshandlung stets an ein Ehlers-Danlos-Syndrom, ein Vitamin-D-Mangel und eine OI (40).
Genetische Beratung
Bei der Vererbung einer OI muss beachtet werden, dass neben dem klassischen dominanten oder rezessiven Erbgang auch irreguläre Übertragungsmuster aufgrund von Mutationen in der Keimbahn resp. der Gonaden mit Entwicklung eines Mosaiks beobachtet werden können (1, 2, 12, 13). Bei dominantem Erbgang beträgt das Wiederholungsrisiko unter der Voraussetzung, dass ein Elternteil Symptome aufweist 50%, bei rezessivem Erbgang 25% und bei gonadalem Mosaik 5–10%. Eine pränatale genetische Diagnostik stellt erhebliche ethische Probleme für Eltern und involvierte Ärzte dar, zumal die Korrelation zwischen Geno- und Phänotyp ungenügend ist, um eine verbindliche Prognose für das individuelle Kind zu erlauben.
Behandlung
Die Ziele der multidisziplinären Behandlung sind eine Reduktion der Frakturhäufigkeit, Verhinderung von Knochendeformierungen und einer Skoliose, Minimierung chronischer Schmerzen und Maximierung der Mobilität und anderer funktioneller Fähigkeiten (2, 14).
Eckstein der medikamentösen Therapie sind Bisphosphonate bei allen OI-Formen mit erhaltener Knochenmineralisation (1, 2, 5, 15–20). Bisphosphonate hemmen die Knochenresorption und den Knochenumsatz und sind im Rahmen der Behandlung der postmenopausalen Osteoporose etabliert. Wenn sie auch für Frakturprophylaxe bei der OI nicht zugelassen sind, zeigte sich in Beobachtungsstudien und einer kontrollierten Studie bei Kindern nach zyklisch verabreichtem Pamidronat (IV, 3-monatlich), der in diesem Zusammenhang am besten dokumentierten Substanz, eine Zunahme der Knochendichte und eine klinisch relevante Reduktion von Frakturen, ohne dass selbst bei kleinen Kindern relevante Nebenwirkungen wie Störungen von Wachstum oder der Frakturheilung aufgetreten wären (5, 15–18, 21, 22). Die günstigen Effekte scheinen in den ersten zwei bis vier Therapiejahren am ausgeprägtesten zu sein, Informationen über die Langzeitverträglichkeit fehlen.
Ähnliche Befunde liegen auch für andere Bisphosphonate vor, allerdings in geringerer Anzahl von Studien und Fällen. Zoledronat sechs- bis zwölfmonatlich intravenös, Risedronat und Alendronat peroral führen zu einer Erhöhung der Knochendichte, wegen kleinen Fallzahlen kann aber nicht immer eine Reduktion der Frakturhäufigkeit objektiviert werden (17, 22–26). Immerhin konnte in einer Vergleichsstudie zwischen Zoledronat und Alendronat eine tiefere klinische Frakturrate unter Zoledronat nachgewiesen werden (27). Da die Kinder noch im Wachstum sind, verbessert sich auch die Wirbelform; es wurde vor allem auch über weniger Knochenschmerzen, eine bessere Mobilität und bessere Muskelkraft berichtet (2, 28).
Die Indikation zu einer Bisphosphonattherapie ist bei Kindern und Erwachsenen mit OI bei Vorliegen von Wirbelfrakturen und gehäuftem Auftreten nicht-vertebraler Frakturen (2 oder mehr Frakturen pro Jahr) gegeben (Tabelle 4). Bei Erwachsenen kann auch ein stark erhöhtes Frakturrisiko allein eine Indikation darstellen.

Die Therapiedauer ist bei der OI weniger gut definiert als bei anderen Formen der Osteoporose. In der Regel wird eine postmenopausale Osteoporose während 3–5 Jahren mit einem Bisphosphonat behandelt (29). Der Grund, weshalb man nicht unbedingt eine Dauertherapie durchführt, ist, dass die Bisphosphonate, die sich ja an den Knochen binden und dort über viele Jahre verweilen, auch nach Absetzen der Behandlung eine Nachwirkung haben. Ein weiterer Grund, dass man heute allenfalls eine Behandlungspause durchführt und dann den Verlauf beobachtet und erst bei einer Verschlechterung der Situation einen erneuten Behandlungszyklus startet, ist, dass man gesehen hat, dass bei Langzeitbehandlungen so genannte atypische Frakturen (Oberschenkelknochen) auftreten und gleichzeitig auch ein gewisses Risiko für eine Osteonekrose des Kieferknochens besteht. Bei der OI wie auch bei den anderen Formen der Osteoporose muss der behandelnde Arzt zusammen mit dem Betroffenen im Einzelnen entscheiden, wie lange die Behandlung durchgeführt werden soll.
Da die Kinder sich im Wachstum befinden und damit der gesamte Knochenumbau deutlich höher ist als bei Erwachsenen, kann man die Therapiedauer nicht einfach in Analogie zu den Erwachsenen festlegen. In Anbetracht der Tatsache, dass die Knochenstoffwechselaktivität bei wachsenden Kindern deutlich höher ist als bei Erwachsenen, kommt bei ihnen oft ein kürzeres Dosierungsintervall zur Anwendung. Für die Dosierung bei Kindern stehen in den behandelnden Institutionen spezielle Dosierungsschemata zur Verfügung.
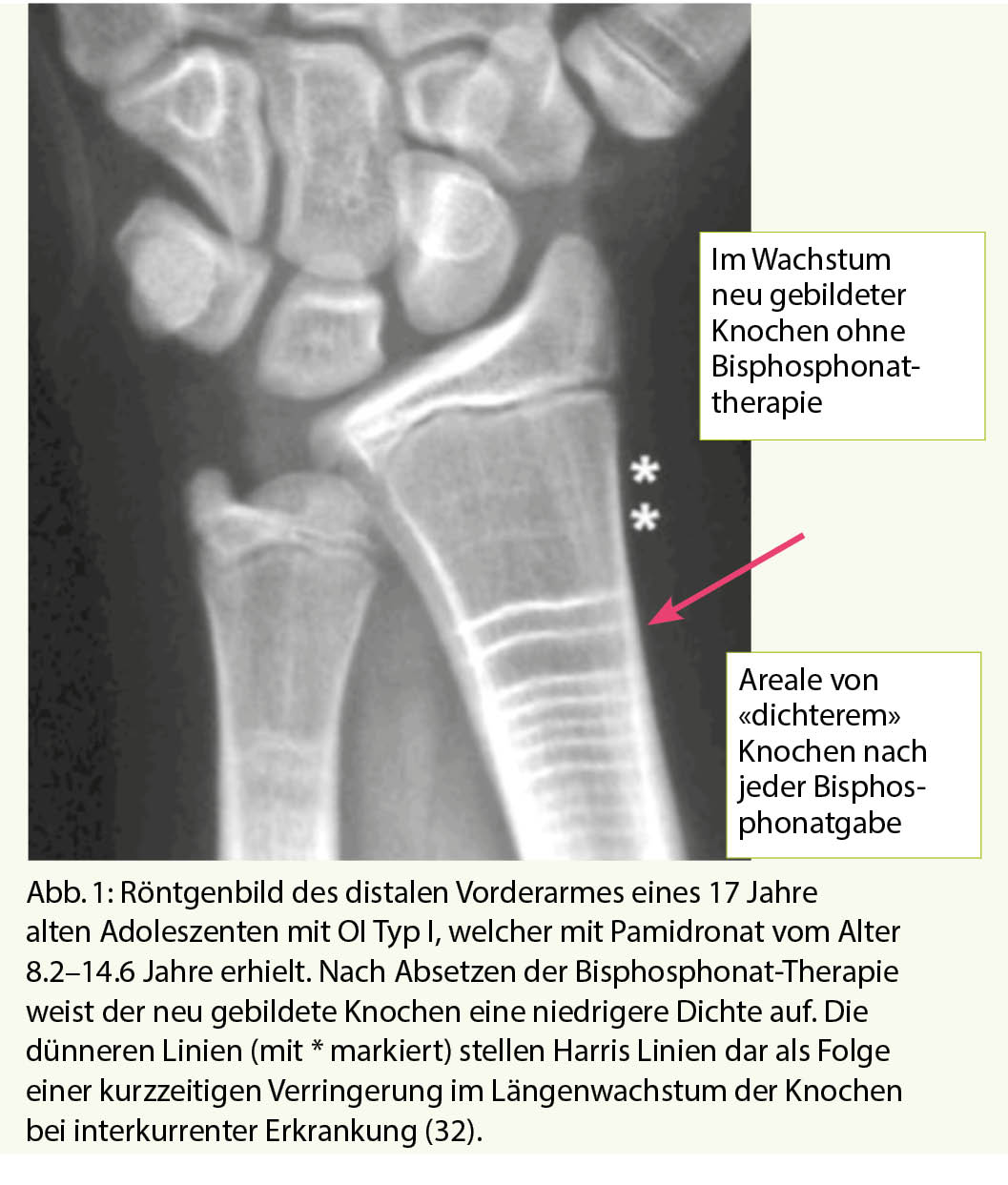
Die Bisphosphonate werden in der Regel zyklisch verabreicht. Im Wachstum ist deshalb der nach der Gabe des Bisphosphonats neu gebildete Knochen nicht oder nur wenig durch das Bisphosphonat beeinflusst und dies ist auch im Röntgenbild sichtbar, indem man Verdichtungslinien sieht (Abbildung 1). Es gibt nun Hinweise, dass der neu gebildete Knochen zwischen dem Bisphosphonat-exponierten Knochen etwas weniger widerstandsfähig ist und es damit bei zu langen Intervallen oder nach Absetzen der Bisphosphonatbehandlung in diesem Bereich vermehrt zu Frakturen kommen könnte (11, 30, 31). Dies hat dazu geführt, dass man neuerdings bei Kindern keine längere Therapiepause vornimmt, sondern die Behandlung weiterführt, aber bei gutem Ansprechen auf die Behandlung die Dosis des Bisphosphonats reduziert. Es ist zum heutigen Zeitpunkt nicht ganz klar, wie lange man dies so weiterführen soll, es scheint aber Sinn zu machen es weiterzuführen, bis die Epiphysenfugen (Wachstumsfugen) geschlossen sind.
Es gibt nun erste Untersuchungen bei einigen OI-Patienten mit Behandlung durch Denosumab. Die ersten Studien beim OI-Typ 6, bei welchem der RANKL eine Rolle spielt, zeigt, dass dieser Typ auf diese Behandlung anspricht (33–35), allerdings ist die Unterdrückung von Osteoklasten von kürzerer Dauer, als bei Individuen mit Osteoporose (35, 36). Im Gegensatz dazu gilt, dass dieser OI-Typ weniger gut auf eine Bisphosphonatbehandlung anspricht als die anderen Typen. Es gibt aber auch neuere Daten, die zeigen, dass das Denosumab auch bei den anderen Typen der OI wirksam scheint (33, 37). Es sind aber noch weitere Studien notwendig, um den Einsatz von Denosumab bei der OI zu evaluieren.
In Analogie zur Behandlung der Osteoporose bei Erwachsenen ist eine begleitende Behandlung mit Calcium und Vitamin D immer empfohlen, ohne dass diese Empfehlung durch spezifische Studien belegt wäre.
Das orthopädische Management von Frakturen der unteren und oberen Extremität sowie der Wirbelsäule gehört in die Hände von Spezialisten. Allgemein gilt, dass Frakturen bei Kleinstkindern primär konservativ behandelt werden, sobald Kinder jedoch zu stehen und gehen beginnen, wird die Stabilisierung der tragenden Knochen und im späteren Leben auch der langen Knochen der Arme mittels Marknagelung zu diskutieren sein. Schrauben- und Platten-osteosynthesen gelten als obsolet, da sie zu gehäuften Frakturen im oberen und unteren Randbereich führen können. Alle chirurgischen Massnahmen müssen von Rehabilitation vor und nach Operation sowie medikamentöser Therapie begleitet werden (Tabelle 5).

Facharzt FMF Innere Medizin und Gastroenterologie
Neuhausstrasse 18
8044 Zürich
Schulthess_hk@swissonline.ch
Klinik für Endokrinologie, Diabetologie und Metabolismus
Universitätsspital Basel
Endonet Praxis und Osteologisches Universitätsforschungszentrum DVO
Aeschenvorstadt 57
4051 Basel
christian.meier@unibas.ch
Speziallabor Hormone und Knochenstoffwechsel
Aeschenvorstadt 57
4051 Basel
marius.kraenzlin@unibas.ch
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
1 Bonafe L, Giunta C, Hasler CC, Janner M, Kraenzlin ME, Link B et al. Osteogenesis imperfecta: Klnik, Diagnose und Management vom Kindes- bis Erwachsenenalter. Schweiz Med Forum 2013; 13(46):925-931.
2 Marini JC, Forlino A, Bachinger HP, Bishop NJ, Byers PH, Paepe A et al. Osteogenesis imperfecta. Nat Rev Dis Primers 2017; 3:17052.
3 Martin E, Shapiro JR. Osteogenesis imperfecta:epidemiology and pathophysiology. Curr Osteoporos Rep 2007; 5(3):91-97.
4 Forlino A, Marini JC. Osteogenesis imperfecta. Lancet 2016; 387(10028):1657-1671.
5 Rauch F, Glorieux FH. Osteogenesis imperfecta. Lancet 2004; 363(9418):1377-1385.
6 Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 2015; 167A(12):2869-2892.
7 Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osterogenesis imperfecta. J Med Genet 1979;(16):-101.
8 Bregou BA, Aubry-Rozier B, Bonafe L, Laurent-Applegate L, Pioletti DP, Zambelli PY. Osteogenesis imperfecta: from diagnosis and multidisciplinary treatment to future perspectives. Swiss Med Wkly 2016; 146:w14322.
9 van Dijk FS, Sillence DO. Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 2014; 164A(6):1470-1481.
10 Rohrbach M, Giunta C. Recessive osteogenesis imperfecta: clinical, radiological, and molecular findings. Am J Med Genet C Semin Med Genet 2012; 160C(3):175-189.
11 Cundy T. Recent advances in osteogenesis imperfecta. Calcif Tissue Int 2012; 90(6):439-449.
12 van Dijk FS, Byers PH, Dalgleish R, Malfait F, Maugeri A, Rohrbach M et al. EMQN best practice guidelines for the laboratory diagnosis of osteogenesis imperfecta. Eur J Hum Genet 2012; 20(1):11-19.
13 van Dijk FS, Dalgleish R, Malfait F, Maugeri A, Rusinska A, Semler O et al. Clinical utility gene card for: osteogenesis imperfecta. Eur J Hum Genet 2013; 21(6).
14 Marr C, Seasman A, Bishop N. Managing the patient with osteogenesis imperfecta: a multidisciplinary approach. J Multidiscip Healthc 2017; 10:145-155.
15 Rauch F, Travers R, Plotkin H, and Glorieux FH. The effects of intravenous pamidronate an the bone tissue of children and adolescents with osteogenesis imprefecta. J Clin Invest 2003; 110:1293-1299.
16 Phillipi CA, Remmington T, Steiner RD. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev 2008;(4):CD005088.
17 Palomo T, Fassier F, Ouellet J, Sato A, Montpetit K, Glorieux FH et al. Intravenous Bisphosphonate Therapy of Young Children With Osteogenesis Imperfecta: Skeletal Findings During Follow Up Throughout the Growing Years. J Bone Miner Res 2015; 30(12):2150-2157.
18 Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev 2016; 10:CD005088.
19 Gatti D, Antoniazzi F, Prizzi R, Braga V, Rossini M, Tato L et al. Intravenous neridronate in children with osteogenesis imperfecta: a randomized controlled study. J Bone Miner Res 2005; 20(5):758-763.
20 Shi CG, Zhang Y, Yuan W. Efficacy of Bisphosphonates on Bone Mineral Density and Fracture Rate in Patients With Osteogenesis Imperfecta: A Systematic Review and Meta-analysis. Am J Ther 2016; 23(3):e894-e904.
21 Rijks EB, Bongers BC, Vlemmix MJ, Boot AM, van Dijk AT, Sakkers RJ et al. Efficacy and Safety of Bisphosphonate Therapy in Children with Osteogenesis Imperfecta: A Systematic Review. Horm Res Paediatr 2015; 84(1):26-42.
22 Lindahl K, Kindmark A, Rubin CJ, Malmgren B, Grigelioniene G, Soderhall S et al. Decreased fracture rate, pharmacogenetics and BMD response in 79 Swedish children with osteogenesis imperfecta types I, III and IV treated with Pamidronate. Bone 2016; 87:11-18.
23 Anam EA, Rauch F, Glorieux FH, Fassier F, Hamdy R. Osteotomy Healing in Children With Osteogenesis Imperfecta Receiving Bisphosphonate Treatment. J Bone Miner Res 2015; 30(8):1362-1368.
24 Bishop N, Adami S, Ahmed SF, Anton J, Arundel P, Burren CP et al. Risedronate in children with osteogenesis imperfecta: a randomised, double-blind, placebo-controlled trial. Lancet 2013; 382(9902):1424-1432.
25 Kumar C, Panigrahi I, Somasekhara AA, Meena BL, Khandelwal N. Zoledronate for Osteogenesis imperfecta: evaluation of safety profile in children. J Pediatr Endocrinol Metab 2016; 29(8):947-952.
26 Idolazzi L, Fassio A, Viapiana O, Rossini M, Adami G, Bertoldo F et al. Treatment with neridronate in children and adolescents with osteogenesis imperfecta: Data from open-label, not controlled, three-year Italian study. Bone 2017; 103:144-149.
27 Lv F, Liu Y, Xu X, Song Y, Li L, Jiang Y et al. ZOLEDRONIC ACID VERSUS ALENDRONATE IN THE TREATMENT OF CHILDREN WITH OSTEOGENESIS IMPERFECTA: A 2-YEAR CLINICAL STUDY. Endocr Pract 2018; 24(2):179-188.
28 Rauch F, Plotkin H, Travers R, Zeitlin L, and Glorieux FH. Osteogenesis Imperfecta Types 1, 3 and 4: Effect of Pamidronate Therapy on Bone and Mineral Metabolism. The Journal of Clinical Endocrinology & Metabolism 2003; 88:986-992.
29 Meier C, Uebelhart B, Aubry-Rozier B, Birkhauser M, Bischoff-Ferrari HA, Frey D et al. Osteoporosis drug treatment: duration and management after discontinuation. A position statement from the SVGO/ASCO. Swiss Med Wkly 2017; 147:w14484.
30 Biggin A, Zheng L, Briody JN, Coorey CP, Munns CF. The long-term effects of switching from active intravenous bisphosphonate treatment to low-dose maintenance therapy in children with osteogenesis imperfecta. Horm Res Paediatr 2015; 83(3):183-189.
31 Biggin A, Briody JN, Ormshaw E, Wong KK, Bennetts BH, Munns CF. Fracture during intravenous bisphosphonate treatment in a child with osteogenesis imperfecta: an argument for a more frequent, low-dose treatment regimen. Horm Res Paediatr 2014; 81(3):204-210.
32 Rauch F, Munns C, Land C, Glorieux FH. Pamidronate in children and adolescents with osteogenesis imperfecta: effect of treatment discontinuation. J Clin Endocrinol Metab 2006; 91(4):1268-1274.
33 Hoyer-Kuhn H, Franklin J, Allo G, Kron M, Netzer C, Eysel P et al. Safety and efficacy of denosumab in children with osteogenesis imperfecta – a first prospective trial. J Musculoskelet Neuronal Interact 2016; 16(1):24-32.
34 Hoyer-Kuhn H, Semler O, Schoenau E. Effect of denosumab on the growing skeleton in osteogenesis imperfecta. J Clin Endocrinol Metab 2014; 99(11):3954-3955.
35 Li G, Jin Y, Levine MAH, Hoyer-Kuhn H, Ward L, Adachi JD. Systematic review of the effect of denosumab on children with osteogenesis imperfecta showed inconsistent findings. Acta Paediatr 2018; 107(3):534-537.
36 Ward L, Bardai G, Moffatt P, Al-Jallad H, Trejo P, Glorieux FH et al. Osteogenesis Imperfecta Type VI in Individuals from Northern Canada. Calcif Tissue Int 2016; 98(6):566-572.
37 Hoyer-Kuhn H, Stark C, Franklin J, Schoenau E, Semler O. Correlation of Bone Mineral Density on Quality of Life in Patients with Osteogenesis Imperfecta during Treatment with Denosumab. Pediatr Endocrinol Rev 2017; 15(Suppl 1):123-129.
38 Mrosk J, Bhavani GS, Shah H, Hecht J, Krüger U et al. Diagnostic strategies and genotype-phenotype correlation in a large Indian cohort of osteogenesis imperfecta. Bone. 2018 May;110:368-377.
39 Palomo T, Vilaça T, Lazaretti-Castro M. Osteogenesis imperfecta: diagnosis and treatment. Curr Opin Endocrinol Diabetes Obes. 2017 Dec;24:381-388.
40 Holick MF, Hossein-Nezhad A, Tabatabaei F. Multiple fractures in infants who have Ehlers-Danlos/hypermobility syndrome and or vitamin D deficiency: A case series of 72 infants whose parents were accused of child abuse and neglect. Dermatoendocrinol. 2017 Feb 16;9:e1279768.
41 Lindert U et al. Urinary pyridinoline cross-links as biomarkers of osteogenesis imperfecta. Orphanet J Rare Dis. 2015; 10: 104.



der informierte @rzt
- Vol. 8
- Ausgabe 7
- Juli 2018