Einführung
Das Ovarialkarzinom steht an Stelle 8 der häufigsten Karzinome der Frau und an Stelle 18 der Krebssterblichkeit. Weltweit wurden 2020 313 000 neue Ovarialkarzinome diagnostiziert (1). In der Schweiz erkranken pro Jahr ca. 600 Frauen an einem Ovarialkarzinom, damit liegt der Anteil an allen Krebsneuerkrankungen bei 3 %. Das mediane Erkrankungsalter liegt bei 68.6 Jahren (2). Die 5-Jahres-Überlebensrate ist mit 46 % eingeschränkt. Trotz Fortschritte in der operativen und systemischen Therapie des Ovarialkarzinoms erleiden die meisten Patientinnen einen Rückfall und versterben schliesslich an ihrer Erkrankung.
In der Karzinogenese spielen reproduktive Faktoren eine Rolle, wie beispielsweise frühe Menarche, späte Menopause, späte Erstgebärende und Nullipara. Risikoreduzierend ist die Einnahme oraler Kontrazeptiva. Weiterhin spielen genetische Faktoren eine grosse Rolle bei der Entstehung des Ovarialkarzinoms (3). Erst seit wenigen Jahren weiss man, dass der Anteil an Patientinnen mit Keimbahnveränderungen in den Genen BRCA1 und BRCA2 bei Patientinnen mit einem sog. high-grade serösen Ovarialkarzinom bei bis zu 20 % liegt, weitere 5 % der Patientinnen haben andere risikoerhöhende Mutationen (z. B. PALB2, RAD 51C) (4). Weitere 20 % der Patientinnen haben eine eingeschränkte DNA-Doppelstrangreparatur (Mangel an homologer Rekombination) ohne das Vorliegen einer pathogenen Mutation (sog. HRD-Positivität) (5).
Früherkennung
Trotz grosser Bemühungen in den vergangenen Jahren existiert weiterhin kein Screening auf Eierstockkrebs bei asymptomatischen Frauen. Der transvaginale Ultraschall (TVUS) gilt als Bildgebungsverfahren der ersten Wahl für die Beurteilung von Ovarialkarzinomen, aber der TVUS allein hat keine ausreichende Sensitivität und Spezifität für die Früherkennung von Eierstockkrebs. Dies ist darauf zurückzuführen, dass das high-grade seröse Ovarialkarzinom seinen mikroskopischen Ursprung in den Eileitern hat und sich bereits früh in der Karzinogenese auf die Eierstöcke und Bauchhöhle ausbreitet. Da eine niedrige Prävalenz den positiven prädiktiven Wert jedes diagnostischen Tests deutlich verringert, besteht das Risiko eines universellen Screenings in falsch-positiven Ergebnissen, die zu Notfällen und diagnostischen Operationen mit potenziell schädlichen Komplikationen führen.
Die UKCTOCS-Studie von Menon et al. war die grösste randomisierte kontrollierte Studie zum Ovarialkarzinom-Screening, an der 200 000 postmenopausale Frauen in Grossbritannien teilnahmen. Ein jährliches multimodales Screening mit einem Algorithmus zur Bestimmung des Tumormarkers CA-125 und/oder einem transvaginalen Ultraschall führten zwar zu einer Verringerung der Diagnose von Eierstockkrebs im fortgeschrittenen Stadium, die krankheitsspezifische Sterblichkeit jedoch verringerte sich nicht (6).
In den letzten Jahren hat die Einführung der opportunistischen bilateralen Salpingektomie als mögliche Strategie zur Verringerung des Risikos von Ovarialkarzinom Einzug gehalten. Die prophylaktische Kastration durch eine bilaterale Salpingo-Oophorektomie bleibt nur Frauen mit genetisch nachgewiesenen BRCA-Genmutationen vorbehalten, da nur hier die Risiken der prämaturen Menopause den Nutzen der Intervention überwiegen (7, 8).
Einteilung
Die meisten Ovarialkarzinome entwickeln sich aus Zellen des Oberflächenepithels; die übrigen entwickeln sich aus anderen Zelltypen (Keimzelltumoren, Keimstrang-Stroma-Tumoren). Selten ist das Ovar Ort von Metastasen (Tab. 1).
Karzinome
Die fünf unterschiedlichen Typen der Karzinome sind biologisch unterschiedlich.
High-grade seröses Karzinom
High-grade seröse Karzinome (HGSC) bilden die grösste Gruppe der Ovarialkarzinome (> 50 % der Fälle). Sie sind aggressive Tumore und werden häufig erst in fortgeschrittenem Stadium entdeckt. Dieser Karzinomtyp ist auch gehäuft mit BRCA1/2-Mutationen assoziiert. Als Ursprungsort der Tumorentstehung wird der Eileiter diskutiert.
Low-grade seröses Karzinom
Low-grade seröse Karzinome sind eine eigene Entität – die Tumorentstehung läuft langsamer als beim HGSC und über Vorstufen von serösen Borderlinetumoren über nicht invasive zu invasiven low-grade serösen Karzinomen (9). Insgesamt haben die low-grade serösen Karzinome zwar eine bessere Prognose, sind jedoch aufgrund schlechter Ansprechraten auf Chemotherapien problematisch. Aufgrund ihrer Seltenheit (5–10 % der Ovarialkarzinome) gab es in den letzten Jahren weniger medikamentöse Therapiefortschritte als beim HGSC. Nebst Chemotherapie haben die antihormonelle Therapie und MEK-Inhibitoren einen wichtigen Stellenwert in aktuellen Behandlungskonzepten.
Muzinöses Karzinom
Diese Karzinome werden häufig im Frühstadium diagnostiziert und entwickeln sich vermutlich aus Zystadenomen oder über Borderlinetumore. Es gibt zwei unterschiedliche Wachstumsmuster: das expansile Muster oder das seltenere, jedoch mit deutlich schlechterer Prognose assoziierte infiltrative Muster. Beim infiltrativen Muster muss eine Metastasierung aus Karzinomen des Gastrointestinaltrakts ausgeschlossen werden.
Endometrioides Karzinom
Endometrioide Karzinome des Ovars entstehen über endometrioide Adenofibrome und Borderlinetumore und werden häufiger in frühen Tumorstadien diagnostiziert. Sie treten häufig in Verbindung mit atypischer Endometriose oder synchron mit einem endometrioiden Karzinom des Corpus uteri auf. Sie werden prinzipiell wie die serösen Ovarialkarzinome behandelt.
Klarzelliges Karzinom
Klarzellige Karzinome treten wie endometrioide Karzinome häufiger bei Frauen mit Endometriose auf und werden immer als high-grade Karzinome klassifiziert. Aufgrund ihrer häufigen Resistenz gegenüber einer platinhaltigen Chemotherapie haben sie in fortgeschrittenen Stadien die schlechteste Prognose aller Ovarialkarzinome. In der Karzinogenese der klarzelligen Ovarialkarzinome spielt die Endometriose, ähnlich wie bei den endometrioiden Ovarialkarzinomen, eine wichtige Rolle.
Maligne Keimzelltumore
Bei dieser Gruppe handelt es sich um die «echten» ovariellen Tumoren, die aus den Keimzellen entstehen. 2/3 aller malignen Ovarialtumore in den ersten zwei Lebensjahrzehnten sind Keimzelltumoren. Der häufigste davon ist das Dysgerminom, gefolgt von gemischten Keimzelltumoren und unreifen Teratomen.
Keimstrang-Stroma-Tumore
Innerhalb der reinen Keimstrangtumore stellen die Granulosazelltumoren (GCTs) die größte Gruppe dar. GCTs sind niedrig-maligne Tumoren, die Östrogene bilden können. Es können Spätrezidive auch noch nach vielen Jahren auftreten. Eine untergeordnete Rolle spielt der Sertoli-Leydig-Zelltumor, der als Hormon Testeron produzieren kann.
Diagnostik
Als bildgebendes Verfahren der ersten Wahl steht der Ultraschall transvaginal zur Verfügung, bei V. a. Karzinom im fortgeschrittenen Stadium sollte ein CT oder eine MRT ergänzend erfolgen. Der V. a. ein Ovarialkarzinom besteht bei Frauen mit unklaren Raumforderungen der Adnexe, unbeabsichtigtem Gewichtsverlust und unklaren abdominalen Beschwerden. Die Symptome sind häufig unspezifisch. Der Tumormarker CA-125 kann als weiteres differenzialdiagnostisches Kriterium herangezogen werden. Gerade bei Frauen nach der Menopause sollten zystische Raumforderungen im Ovar beobachtet bzw. histologisch gesichert werden. Bei Frauen im reproduktionsfähigen Alter sind die meisten zystischen Befunde funktionelle Zysten. Eine Raumforderung im Unterbauch mit Aszites deutet in der Regel auf ein Ovarialkarzinom hin. Die diagnostische Laparoskopie zur histologischen Sicherung und Beurteilung einer Operabilität stellt eine Option, vor allem bei fortgeschrittenen Stadien, dar (10).
Stadieneinteilung
Die Stadieneinteilung des Ovarialkarzinoms erfolgt als operatives Staging nach FIGO (Tab. 2) und bestimmt die Radikalität der Operation sowie die adjuvanten Therapien.

Die Behandlung des Ovarialkarzinoms richtet sich nach Stadium, Grad und Histologie und basiert auf drei Säulen: operative Zytoreduktion, Chemotherapie und zielgerichtete Erhaltungstherapie. Gemäss der neuen schweizerischen IVHSM-Richtlinie werden Ovarialkarzinomoperationen in Zukunft zur hoch spezialisierten Medizin gehören und auf dafür ausgewählte Zentren limitiert werden.
Folgende Grundprinzipien gelten:
– Im frühen Tumorstadium (Stadium IA oder IB) und/oder bei endometrioiden Tumoren des Grades 1 ist nach einer alleinigen Operation die Prognose ausgezeichnet (Überlebensrate 90 %).
– Ab Stadium IC, II, Grad 3 oder klarzelliger Histologie wird eine adjuvante Chemotherapie (z. B. mit Carboplatin und Paclitaxel) empfohlen.
– Im Stadium III oder IV ist die primäre operative Zytoreduktion, gefolgt von einer systemischen Chemotherapie, die Standardbehandlung. Kommt eine Patientin aufgrund der Tumorausdehnung oder aufgrund von Begleiterkrankungen für eine primäre Operation nicht infrage, ist eine neoadjuvante Chemotherapie, gefolgt von einer zytoreduktiven Operation, eine Alternative.
– Bei Frauen mit Kinderwunsch im Stadium IA kann ein fertilitätserhaltendes Vorgehen (Erhalt von einem Eierstock und dem Uterus) erwogen werden.
Therapie
Operative Therapie
Generell sollte die operative Behandlung des Ovarialkarzinoms durch gynäkologische Onkologen an einem zertifizierten Tumorzentrum erfolgen.
Frühstadium
Bei Verdacht auf ein Karzinom im Frühstadium erfolgt das Staging und die Stadieneinteilung chirugisch (StagingOperation). Diese kann mittels Laparoskopie oder robotergeführter laparoskopischer Chirurgie erfolgen (12). Im Regelfall ist jedoch eine Laparotomie erforderlich, die einen guten Zugang zum Oberbauch und damit eine korrekte Abschätzung der Tumorausdehnung ermöglicht. Die Operation umfasst die Hysterektomie und die bilaterale Salpingo-Oophorektomie. Alle peritonealen Oberflächen, die Zwerchfellhälften und die Organe des Abdomens und des Beckens werden beurteilt. Es wird Spülflüssigkeit vom Becken (Douglas-Raum) gewonnen, und multiple Peritonealbiopsien werden im mittleren und lateralen Becken und Abdomen entnommen. In frühen Tumorstadien wird zu Staging-Zwecken das infrakolische Omentum reseziert und eine pelvine und paraaortale Lymphadenektomie durchgeführt. Eine Sentinel-Lymphknoten-Biopsie wird bei Patientinnen mit Ovarialkarzinom aktuell in klinischen Studien erforscht. Anzumerken ist, dass die Entscheidung für den operativen Zugangsweg nicht auf Kosten der onkologischen Sicherheit stattfinden sollte.
Fortgeschrittenes Stadium
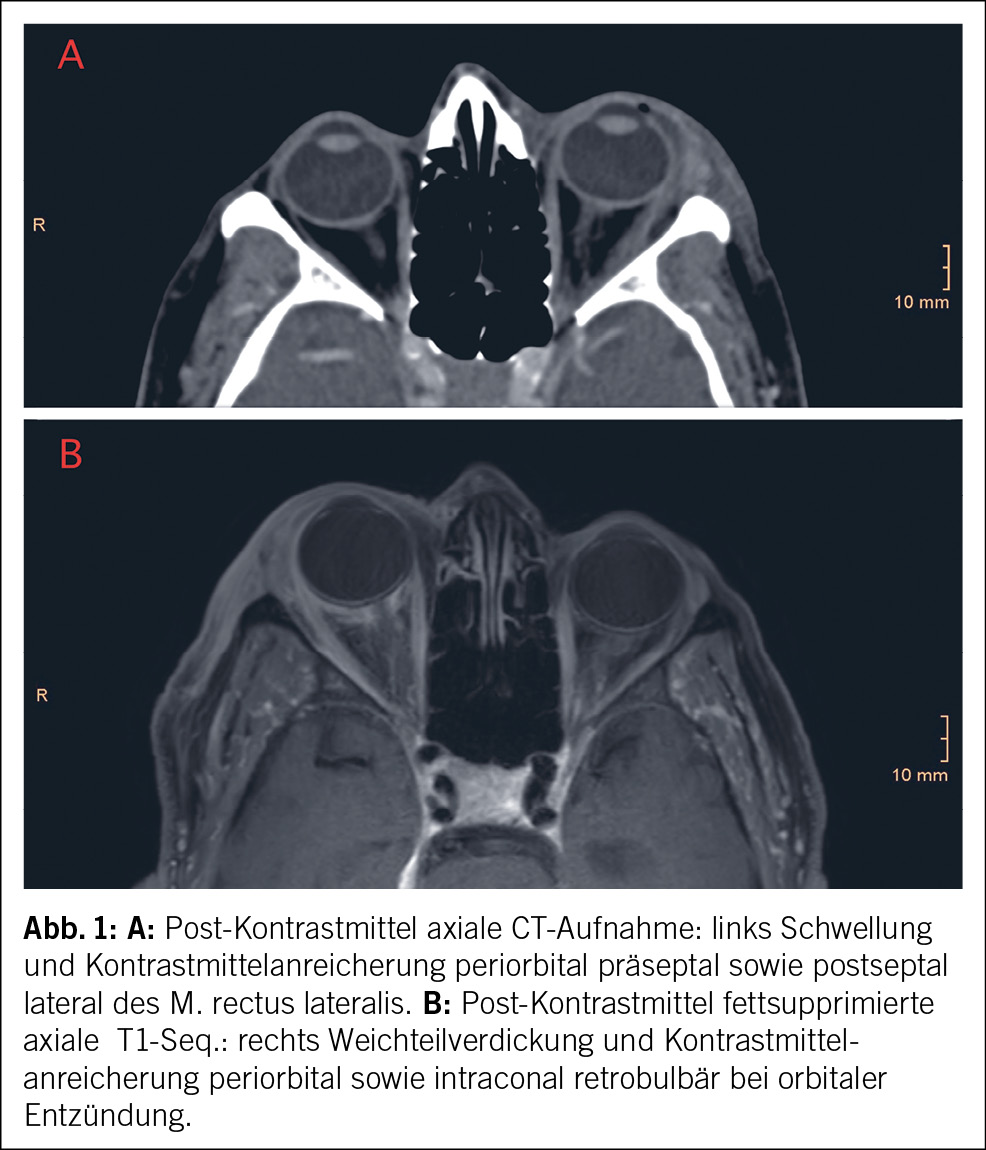
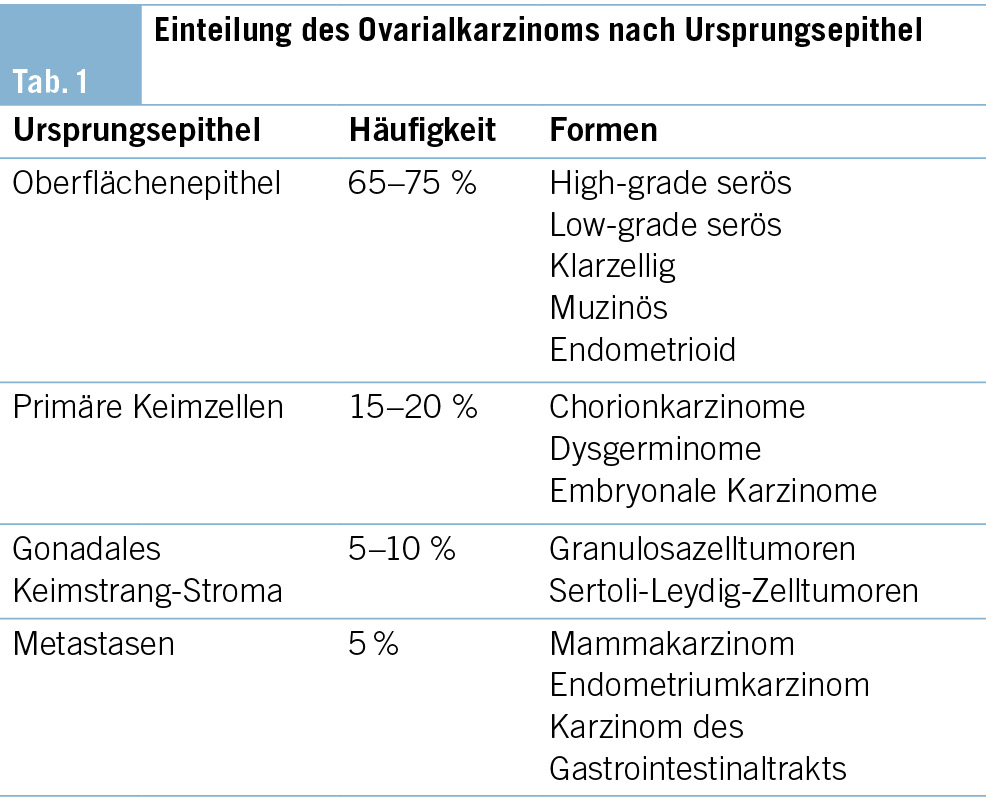
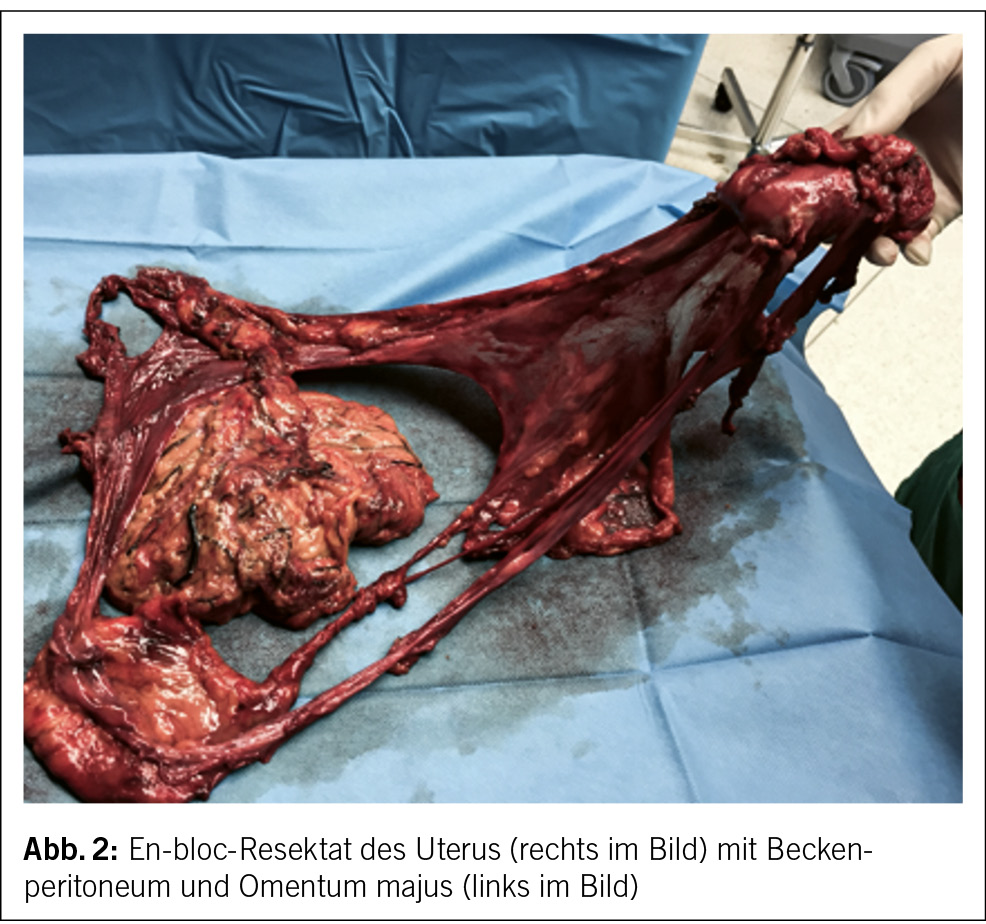
Die komplette Tumorentfernung ist das primäre Ziel der Operation beim fortgeschrittenen Ovarialkarzinom (Debulking-Operation) (Abb. 1 und 2). Diese umfasst neben der Hysterektomie mit Salpingo-Oophorektomie bds. die Resektion von allen sichtbaren Tumorabsiedlungen im Bauchraum. In der Regel werden eine Resektion des Zwerchfellperitoneums, eine Entfernung von Peritoneum im kleinen Becken und in den Kolonrinnen, die infragastrische Omentektomie und ggf. Darmresektionen, Leberteilresektionen, Splenektomien etc. durchgeführt. Die systematische pelvine und paraaortale Lymphondektomie wird nur beim Tumorbefall der regionalen Lymphknoten durchgeführt. Eine grosse multizentrische, randomisierte europäische Studie zeigte bei fortgeschrittenen Ovarialkarzinomen ab dem Stadium FIGO-IIB keinen Überlebensvorteil für die systematische Lymphonodektomie bei klinisch unauffälligen Lymphknoten, nur eine erhöhte Morbidität (13). Die Zytoreduktion auf keine sichtbare Erkrankung mehr (makroskopische Tumorfreiheit) verbessert das Überleben der Patientinnen. Sollte eine komplette Zytoreduktion nicht erreicht werden, wird versucht, einen verbleibenden Tumorrest unter 1 cm zu erreichen.
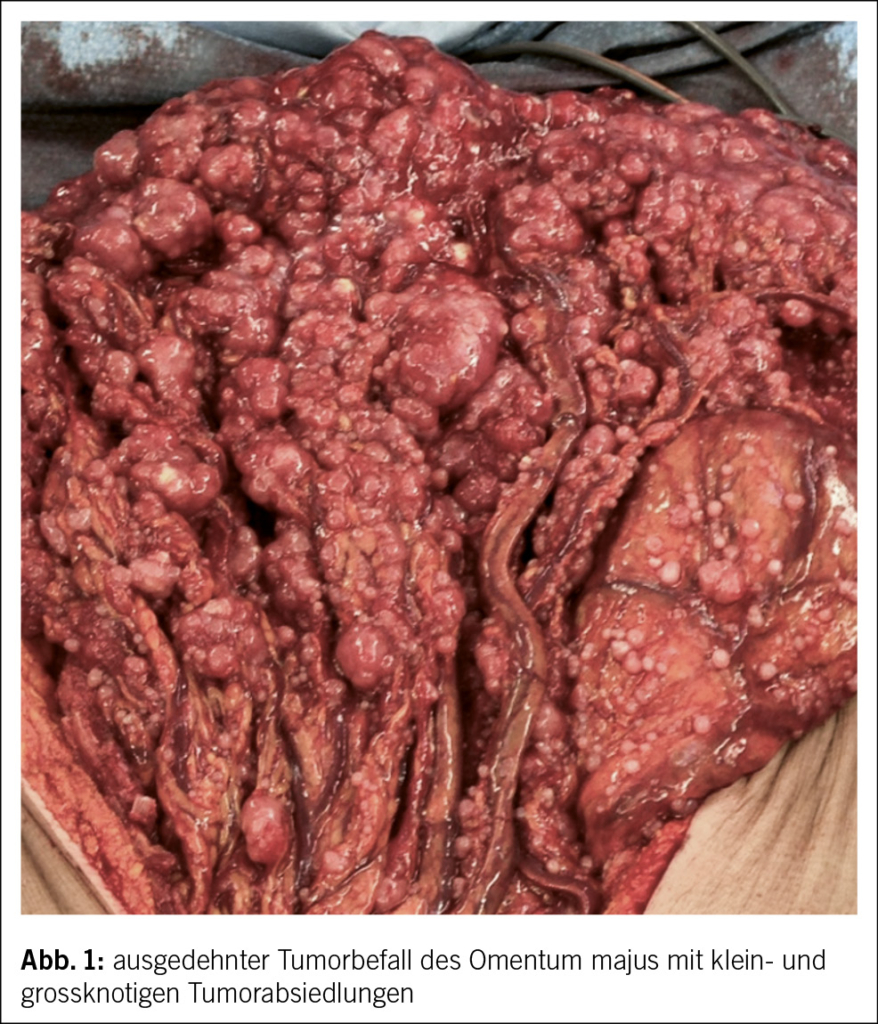
Liegt doch ein inoperabler Situs vor, d.h., es ist keine komplette Resektion möglich mit verbleibendem Tumorrest über 1 cm Durchmesser der einzelnen Metastasen, profitieren die Betroffenen nicht mehr von der chirurgischen Resektion. Hier sollte eine primäre Systemtherapie erfolgen. Um unnötige chirurgische Interventionen zu vermeiden, hat die diagnostische Laparoskopie in den letzten Jahren an Stellenwert gewonnen. Der laparoskopisch ermittelte Fagotti-Score evaluiert die Operabilität durch eine Beurteilung der Tumorlast im Abdomen und ist der in Europa am meisten verwendete Score. Bei Patientinnen mit einem hohen Fagotti-Score ist eine optimale primäre Zytoreduktion sehr unwahrscheinlich. Diese Patientinnen können zunächst eine neoadjuvante Chemotherapie erhalten, bei Ansprechen dann gefolgt von einer Intervalloperation. Es kann aber auch sein, dass ein reduzierter Allgemeinzustand ein primäres Tumordebulking nicht ermöglicht.
Systemische Therapie
Es gibt in der Primärbehandlung des Ovarialkarzinoms prinzipiell zwei Möglichkeiten der systemischen Therapie:
– Staging- oder Debulking-Operation, gefolgt von 6 Zyklen einer platinhaltigen Chemotherapie und ggf. Erhaltungstherapie
– 3 Zyklen neoadjvuanter Chemotherapie, gefolgt von einer Operation bei Ansprechen auf die Therapie und Gabe von 3 weiteren Chemotherapiezyklen postoperativ und ggf. Erhaltungstherapie
Die Standardchemotherapie besteht aus 6 Zyklen Paclitaxel und Carboplatin. Für bestimmte Patientinnen wird der Antiangiogenese-Antikörper Bevacizumab hinzugefügt und als Erhaltungstherapie fortgeführt. In einer multizentrischen, randomisierten Phase-III-Studie wurde die Applikationsdauer auf 15 Monate festgelegt (im Vergleich zu 30 Monaten) (14).
Bei Patientinnen mit BRCA1- und/oder BRCA2-Mutationen ist der Einsatz des PARP-Inhibitors Olaparib durch die SOLO1/GOG-3004-Studie belegt. Patientinnen ab FIGO-Stadium-III mit high-grade serösem oder endometrioidem Ovarialkarzinom erhielten bei Ansprechen nach Abschluss der platinhaltigen Chemotherapie eine 2-jährige Erhaltungstherapie mit Olaparib (300 mg 2 x täglich). Nach 7 Jahren lebten 67 % der Patientinnen, die Olaparib erhalten hatten, und nur 46.5 % der Patientinnen mit Placebo, davon waren 45.3 % bzw. 20.6 % der Patientinnen ohne Rezidiv (15).
Die Möglichkeit der Kombination von Bevacizumab und einem PARP-Inhibitor wurde in der PAOLA-1/ENGOT-ov2-Studie überprüft. Patientinnen mit einer BRCA1- oder BRCA2-Mutation oder einem positiven HRD-Score ab FIGO-Stadium-III mit high-grade serösem oder endometrioidem Ovarialkarzinom erhielten bei Ansprechen nach Abschluss der platinhaltigen Chemotherapie eine 2-jährige Erhaltungstherapie mit Bevacizumab plus Olaparib. Das progressionsfreie Überleben verlängerte sich durch Olaparib von 21.7 auf 37.2 Monate bei Vorliegen einer BRCA1/2- Mutation und von 16.6 auf 28.1 Monate in der HRD-positiven Gruppe ohne Mutation. HRD-Positivität (Score >= 42) wurde mittels eines kommerziell erhältlichen Tests am Tumorgewebe (MyChoice® von Myriad genetics) bestimmt, was die Übertragbarkeit in die Routine erschwert. In der Schweiz ist an der Universitätsklinik Genf (HUG) ein akademischer HRD-Test erhältlich, welcher auf der PAOLA-I-Studienpopulation retrospektiv validiert wurde. Weitere kommerzielle HRD-Tests sind ebenfalls erhältlich (z. B. Foundation-One® von Foundation Medicine). Ob eine zusätzliche endokrine Therapie mit dem Aromatasehemmer Letrozol für 2 Jahre eine Option sein könnte, wird in der aktuell rekrutierenden ENGOT-ov54/Swiss-GO-2/MATAO-Studie untersucht.
Die hyperthermische intraperitoneale Chemotherapie (HIPEC), welche im Rahmen einer Intervall-Debulking-Operation bei einer makroskopischen Komplettresektion einen möglichen Stellenwert haben könnte, bleibt aufgrund widersprüchlicher Studienresultate weiterhin sehr umstritten und sollte auch aufgrund der vermehrten Nebenwirkungen nur im Rahmen von klinischen Studien appliziert werden (16). Dies gilt insbesondere auch in der Rezidivsituation, wo eine HIPEC nach sekundärer Debulking-Operation bisher keinen Vorteil aufzeigen konnte. Auch damit verwandte Therapiekonzepte wie die intraperitoneale Hochdruck-Aerosol-Chemotherapie (PIPAC) befinden sich zurzeit noch in experimenteller Entwicklung (17).
Nachsorge
Nach der abgeschlossenen Primärtherapie, noch während der Durchführung einer Erhaltungstherapie, wird eine regelmässige gynäkologisch-onkologische Nachsorge durchgeführt. Ziel der Nachsorge sind die Erkennung von therapieassoziierten Nebenwirkungen, die psychosoziale Betreuung, die Verbesserung der Lebensqualität und die Erkennung des Rezidivs. Die wissenschaftliche Evidenz zur Relevanz der Nachsorge ist limitiert. Die Nachsorge soll eine sorgfältige Anamneseerhebung, die körperliche Untersuchung inklusive gynäkologischer Spiegel- und Tastuntersuchung und die Vaginalsonographie sowie orientierende abdominale Sonographie umfassen. In den ersten 3 Jahren nach Abschluss der Therapie sollte die Untersuchung in 3-monatlichen Intervallen, im 4. und 5. Jahr in 6-monatlichen Intervallen und danach halbjährlich bis jährlich erfolgen. Während einer laufenden Erhaltungstherapie mit Bevacizumab oder einem PARP-Inhibitor sollte alle 3 Monate eine Tumormarkerkontrolle (CA-125) zusätzlich durchgeführt werden. Im Anschluss an die Erhaltungstherapie sollte eine Tumormarkerkontrolle nur bei Symptomen bzw. bei V. a. Rezidiv durchgeführt werden und auch nur bei V. a. Rezidiv eine bildgebende Untersuchung (CT oder MRT) indiziert werden. Eine Mammadiagnostik wird zweijährlich empfohlen.
Zur Sicherheit einer Hormonersatztherapie kann keine zuverlässige Aussage gemacht werden, kann aber nach entsprechender Aufklärung durchgeführt werden.
Rezidiv
Im Falle eines Tumorrezidivs entscheiden der zeitliche Abstand zur vorherigen abgeschlossenen Chemotherapie, das Ausmass der Erkrankung sowie der Allgemeinzustand der Patientin über die weitere Therapie.
In einigen Fällen kann eine zweite Operation im Rezidiv durchgeführt werden. In jedem Fall erfolgt eine erneute Chemotherapie als Kombinationstherapie mit einem Platin oder als Monotherapie. Auch neuere Therapieoptionen mit Antibody-Drug-Konjugaten werden in dieser Therapiesituation in Studien evaluiert. Immuntherapien (Checkpointinhibitoren) haben im Vergleich zu vielen anderen Entitäten beim Ovarialkarzinom bislang zu keinem wesentlichen Behandlungsfortschritt geführt.
Historie
Manuskript eingereicht: 12.09.2024
Angenommen nach Revision: 07.01.2025
Klinik für Gynäkologie
Universitätsspital Zürich (USZ)
isabell.witzel@usz.ch
Die Autorinnen und Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Ovarian Cancer Statistics: World Cancer Research Fund International; 2022 Available from: https://www.wcrf.org/cancer-trends/ovarian-cancer-statistics/.
2. Krebs, Neuerkrankungen und Sterbefälle: Anzahl, Raten, Medianalter und Risiko pro Krebsart: Bundesamt für Statistik / Nationale Krebsregistrierungsstelle; 12.12.2023 [Available from: https://www.bfs.admin.ch/bfs/de/home/statistiken/gesundheit/gesundheitszustand/krankheiten/krebs/indikatoren-arten.assetdetail.29145337.html.
3. Havrilesky LJ, Abernethy AP. Quality of life in ICON7: need for patients‘ perspectives. Lancet Oncol. 2013;14(3):183-5.
4. Harter P, Hauke J, Heitz F, Reuss A, Kommoss S, Marmé F, et al. Prevalence of deleterious germline variants in risk genes including BRCA1/2 in consecutive ovarian cancer patients (AGO-TR-1). PLOS ONE. 2017;12(10):e0186043.
5. Pujade-Lauraine E, Brown J, Barnicle A, Wessen J, Lao-Sirieix P, Criscione SW, et al. Homologous Recombination Repair Gene Mutations to Predict Olaparib Plus Bevacizumab Efficacy in the First-Line Ovarian Cancer PAOLA-1/ENGOT-ov25 Trial. JCO Precis Oncol. 2023;7:e2200258.
6. Menon U, Gentry-Maharaj A, Burnell M, Singh N, Ryan A, Karpinskyj C, et al. Ovarian cancer population screening and mortality after long-term follow-up in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. The Lancet. 2021.
7. Marchetti C, De Felice F, Palaia I, Perniola G, Musella A, Musio D, et al. Risk-reducing salpingo-oophorectomy: a meta-analysis on impact on ovarian cancer risk and all cause mortality in BRCA 1 and BRCA 2 mutation carriers. BMC Women‘s Health. 2014;14(1).
8. Finch AP, Lubinski J, Moller P, Singer CF, Karlan B, Senter L, et al. Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J Clin Oncol. 2014;32(15):1547-53.
9. Lazurko C, Linder R, Pulman K, Lennox G, Feigenberg T, Fazelzad R, et al. Bevacizumab Treatment for Low-Grade Serous Ovarian Cancer: A Systematic Review. Curr Oncol. 2023;30(9):8159-71.
10. Fagotti A, Ferrandina G, Fanfani F, Ercoli A, Lorusso D, Rossi M, et al. A laparoscopy-based score to predict surgical outcome in patients with advanced ovarian carcinoma: a pilot study. Ann Surg Oncol. 2006;13(8):1156-61.
11. Berek JS, Renz M, Kehoe S, Kumar L, Friedlander M. Cancer of the ovary, fallopian tube, and peritoneum: 2021 update. International Journal of Gynecology & Obstetrics. 2021;155(S1):61-85.
12. Gallotta V, Petrillo M, Conte C, Vizzielli G, Fagotti A, Ferrandina G, et al. Laparoscopic Versus Laparotomic Surgical Staging for Early-Stage Ovarian Cancer: A Case-Control Study. J Minim Invasive Gynecol. 2016;23(5):769-74.
13. Harter P, Sehouli J, Lorusso D, Reuss A, Vergote I, Marth C, et al. A Randomized Trial of Lymphadenectomy in Patients with Advanced Ovarian Neoplasms. New England Journal of Medicine. 2019;380(9):822-32.
14. Pfisterer J, Joly F, Kristensen G, Rau J, Mahner S, Pautier P, et al. Optimal treatment duration of bevacizumab (BEV) combined with carboplatin and paclitaxel in patients (pts) with primary epithelial ovarian (EOC), fallopian tube (FTC) or peritoneal cancer (PPC): A multicenter open-label randomized 2-arm phase 3 ENGOT/GCIG trial of the AGO Study Group, GINECO, and NSGO (AGO-OVAR 17/BOOST, GINECO OV118, ENGOT Ov-15, NCT01462890). Journal of Clinical Oncology. 2021;39(15_suppl):5501-.
15. DiSilvestro P, Banerjee S, Colombo N, Scambia G, Kim BG, Oaknin A, et al. Overall Survival With Maintenance Olaparib at a 7-Year Follow-Up in Patients With Newly Diagnosed Advanced Ovarian Cancer and a BRCA Mutation: The SOLO1/GOG 3004 Trial. J Clin Oncol. 2023;41(3):609-17.
16. Van Driel WJ, Koole SN, Sikorska K, Schagen Van Leeuwen JH, Schreuder HWR, Hermans RHM, et al. Hyperthermic Intraperitoneal Chemotherapy in Ovarian Cancer. New England Journal of Medicine. 2018;378(3):230-40.
17. Vergote I, Harter P, Chiva L. Is There a Role for Intraperitoneal Chemotherapy, Including HIPEC, in the Management of Ovarian Cancer? J Clin Oncol. 2019;37(27):2420-3.