Le pronostic des patientes atteintes d’ un cancer du sein s’ est sensiblement amélioré lors des dernières années. En Europe, le taux de mortalité du cancer du sein a diminué (selon les estimations) de 8,7 % entre 2014 et 2019 (1). Le Dr méd. Konstantin Dedes et le Dr méd. Christian Kurzeder, privat-docents, fournissent dans cette interview des éléments de réponse concernant les facteurs ayant contribué à cette évolution positive.
Le pronostic des patientes atteintes d’ un cancer du sein s’ est sensiblement amélioré lors des dernières années. En Europe, le taux de mortalité du cancer du sein a diminué (selon les estimations) de 8,7 % entre 2014 et 2019 (1). Le Dr méd. Konstantin Dedes et le Dr méd. Christian Kurzeder, privat-docents, fournissent dans cette interview des éléments de réponse concernant les facteurs ayant contribué à cette évolution positive.
Die venöse Thromboembolie (VTE) ist ein multifaktorielles Geschehen. Gemäss Virchow verursacht durch einen beeinträchtigten Blutfluss (Stase), ein Gefässtrauma oder eine Hyperkoagulabilität. Ob und wie eine Gerinnungsabklärung im Management einer VTE hilft, soll im Folgenden beleuchtet werden.
Die Inzidenz der venösen Thromboembolie ist nicht wesentlich anders als jene des Myokardinfarkts 1-2/1000 Personen. Diese steigt deutlich mit dem Alter. Wegen Schwangerschaft, Wochenbett, hormoneller Verhütung und Unfällen sind aber jüngere Personen nicht unwesentlich betroffen. In Europa dürften jährlich 500 000 Todesfälle in Zusammenhang mit venösen Thromboembolien stehen (1). Für über 80% der aufgetretenen Thromboembolien lassen sich Risikofaktoren finden (Tab. 1) (2, 3). Mehr als 2/3 der Ereignisse treten provoziert durch Operationen, Hospitalisation und Tumorleiden auf. Primär gilt es, Thromboembolien durch geeignete prophylaktische Massnahmen zu verhindern. Tritt eine VTE auf müssen Symptome rasch richtig gedeutet, die Diagnose bildgebend gesichert und die Behandlung eingeleitet werden. Mit Ausnahme des Nachweises von Antiphospholipid Antikörpern und Lupus Antikoagulans beeinflusst kein Gerinnungstest die Art der Behandlung. In der akuten Situation einer VTE hat die Behandlung oberste Priorität. Eine streng systematische Herangehensweise mit Anamnese, körperlicher Untersuchung und Übersichtslabors helfen die aufgetretene VTE richtig einzuordnen. Eine Gerinnungsabklärung steht nur bei selektionierten Patienten am Ende dieses Prozesses.
Systematischer Ansatz zur Abklärung venöser Thromboembolie
Anamnese: Nach Risiken wie operativen Eingriffen, Traumata und Hospitalisationen (bis 90 Tage zurück!), Immobilisationen (Reiseanamnese), Schwangerschaften ist zu fragen. Frühere Thromboembolien und Erkrankungen mit erhöhtem Thromboserisiko wie systemischer Lupus erythematodes, myeloproliferative Erkrankungen, nephrotisches Syndrom, entzündliche Darmerkrankungen müssen in Erfahrung gebracht werden. Eingenommene Medikamente, besonders hormonelle Kontrazeptiva, Hormonersatzbehandlungen, Testosteronsubstitution, Tamoxifen und Steroidanwendung sind festzuhalten. Frauen sind nach Aborten zu fragen. Die Familienanamnese ist hinsichtlich Thromboembolien zu erfassen. Nach Symptomen, welche auf eine Neoplasie hinweisen könnten, wie Gewichtsabnahmen, Müdigkeit, Appetitverlust, Husten, Hämoptoe, Stuhlunregelmässigkeiten, Hämaturie ist gezielt zu fragen. Status: Bei der körperlichen Untersuchung sucht man Veränderungen, welche auf eine maligne Erkrankung hinweisen könnten wie Lymphadenopathie, Verhärtungen der Mamma oder Testes, Aszites, Hepatomegalie, Splenomegalie, Ödeme. Übersichtslabor: Blutbild mit Ausstrich für mikroskopische Beurteilung, Gerinnungsstatus inkl. Lupus Antikoagulans Test, CRP/BSR, Leber- und Nierenwerte, Urinstatus und allenfalls Hämoccult-Test. Pathologischen Befunden ist nachzugehen. Bildgebung: Die Bildgebung muss die Diagnose der Thromboembolie sichern und soll die Lokalisation und Ausdehnung der Gerinnsel festhalten.
Thrombophilieabklärung für wen?
Es besteht allgemeiner Konsens, dass nicht jede Person mit einer durchgemachten Thromboembolie eine Thrombophilieabklärung braucht (4, 5). Denn der Nachweis einer Thrombophilie beeinflusst die Therapie und spätere Prophylaxe kaum. Die Mortalität ändert nicht. Die am häufigsten anzutreffenden Thrombophilien heterozygote Faktor-V-Leiden-Mutation und heterozygote Prothrombinmutation beeinflussen das Rezidiv-
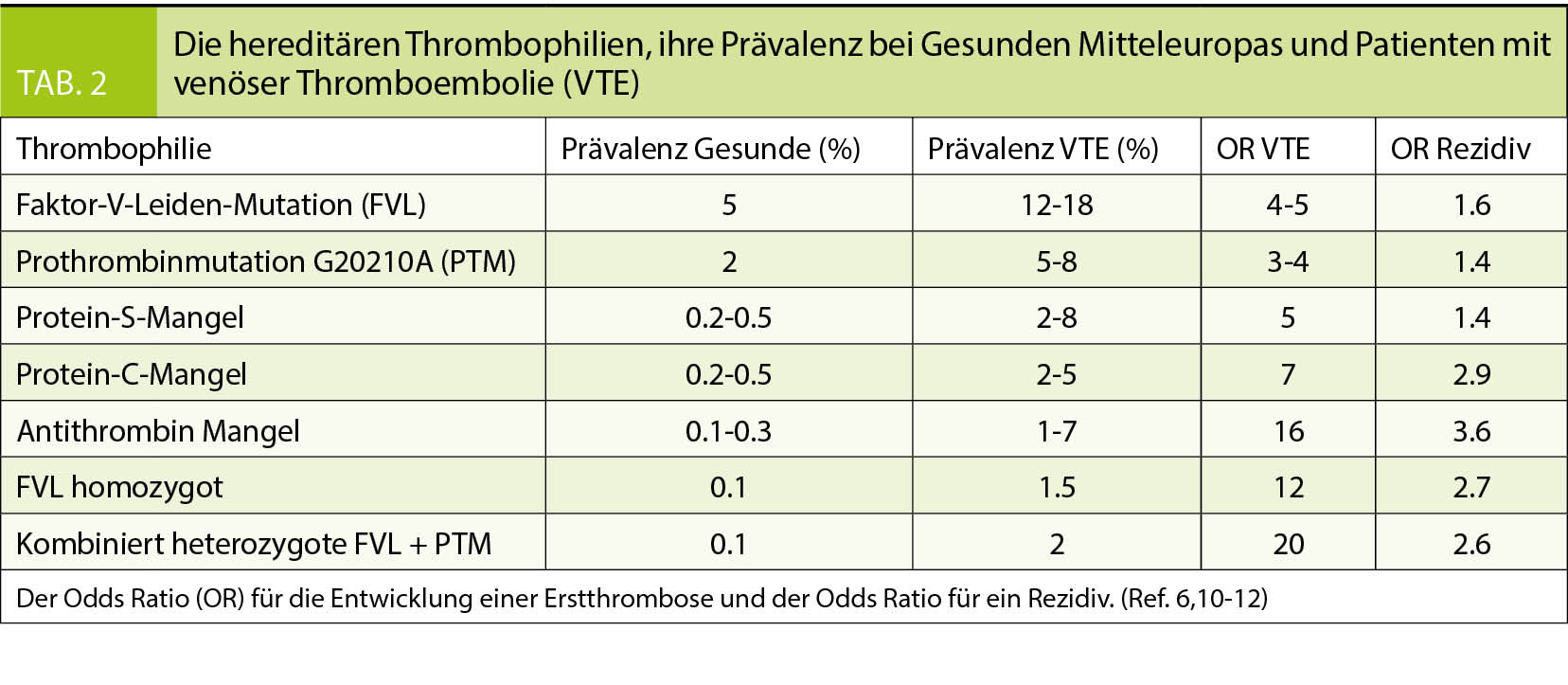
risiko nach Erstthrombose nicht. Die Thrombophilien, welche die Rückfallgefahr und damit die Dauer der Antikoagulation beeinflussen könnten (Antithrombin-, Protein-C-Mangel, homozygote Faktor-V-Leiden-Mutation, kombiniert heterozygote Faktor-V-Leiden plus heterozygote Prothrombinmutation) sind selten (Tab. 2). In folgenden Situationen ist die Gerinnungsuntersuchung aber empfohlen: Positive Familienanamnese: Hat eine oder haben mehrere direkt verwandte Personen (Eltern, Geschwister, Kinder) im Alter unter 45 Jahre eine Thromboembolie durchgemacht, ist die Wahrscheinlichkeit deutlich erhöht, dass bei der Abklärung eine Thrombophilie gefunden wird (6). Eine hereditäre Thrombophilie ist zu suchen. Junge Patienten unter 45-50 Jahre: Patienten mit einer Thrombo-
embolie in dieser Altersgruppe haben in bis zur Hälfte der Fälle eine Thrombophilie. Sie sollen auf das Vorliegen einer angeborenen Thrombophilie und ein Antiphospholipid Syndrom untersucht werden. Patienten mit wiederkehrenden Thromboembolien: Bei wiederkehrenden Thromboembolien in relativ kurzer Folge ist nach einer angeborenen Thrombophilie und einem Antiphospholipid Syndrom zu suchen. Die Schwelle, eine okkulte Neoplasie zu suchen wird niedrig. Patienten mit atypisch lokalisierten Thrombosen: Patienten mit Pfortader-, Lebervenen-, Mesenterialvenen- und Cerebralvenenthrombosen sind auf eine hereditäre Thrombophilie und ein Antiphospholipid Syndrom zu testen. Bei splanchnischen Thrombosen liegt mit recht hoher Wahrscheinlichkeit ein myeloproliferatives Syndrom (MPS) vor. Man sucht eine JAK-2 V617F Mutation. Daneben ist auch an eine paroxysmale nächtliche Hämoglobinurie (PNH) zu denken. In der Regel ist in diesen speziellen Situationen eine Vorstellung beim Hämatologen ratsam. Patienten mit arteriellen Thrombosen: Bei arteriellen Thrombosen kann ein Antiphospholipid Syndrom wesentliche Ursache sein und sollte besonders bei jüngeren Patienten gesucht werden. Besteht auch eine Möglichkeit für gekreuzte Embolien (offenes Foramen ovale) ist auch eine hereditäre Thrombophilie zu suchen. Andernfalls sind diese nicht Risiken für arterielle Thrombosen.
Nutzen und potentieller Schaden der Thrombophilieabklärung bei diesen selektionierten Patienten
Der Nachweis einer Thrombophilie kann in zukünftigen Risiko-situationen zu einer personalisierten Thromboseprophylaxe benutzt werden, die dosisintensiviert und verlängert durchgeführt wird. Der Nachweis einer hereditären Thrombophilie kann helfen, Verwandte ersten Grades mit erhöhtem Thromboserisiko zu finden. Alle hereditären Thrombophilien werden autosomal dominant vererbt. Direkt Verwandte haben deshalb eine Vortestwahrscheinlichkeit von 50% die bekannte Thrombophilie auch aufzuweisen. Die Kenntnis, dass eine Gefährdung für Thrombosen vorliegt, kann dazu verwendet werden, Hormone gezielt und restriktiv einzusetzen und Thromboseprophylaxen individualisiert durchzuführen. Das Wissen um einen Antithrombin Mangel z.B. könnte verwendet werden, in Risikosituationen wie z.B. während einer Schwangerschaft eine Thromboseprophylaxe und peripartal eine Antithrombinsubstitution durchzuführen.
Mögliche Nachteile
Da der Nachweis einer Thrombophilie keinen Überlebensvorteil bringt, ist er primär mit Kosten und wenig belegtem Nutzen verbunden. Der Nachweis einer Thrombophilie könnte zu einer unreflektiert langen Antikoagulation führen. Die Diagnose Thrombophilie kann in den Betroffenen Ängste provozieren. Der Patient ist deshalb in den Entscheid eine Abklärung durchzuführen einzubeziehen.
Thrombophilieabklärung, wenn ja, was zu welchem Zeitpunkt?
Ein Antiphospholipid Syndrom kann dann diagnostiziert werden wenn klinische Ereignisse wie anderweitig nicht erklärte venöse und arterielle Thromboembolien, wiederholte Aborte, intrauteriner Fruchttod, schwere Präeklampsie oder eine Plazentainsuffizienz mit dem Nachweis von Laborbefunden wie erhöhten IgG/IgM-Titern von Anti-Cardiolipin und/oder Anti-β2-Glykoprotein-1-Antikörpern und/oder einem positiven Lupus-Antikoagulans Test zusammentreffen. Die Laborbefunde müssen im Abstand von mindestens 12 Wochen zweimal positiv ausfallen bevor die Diagnose Antiphospholipid Syndrom gestellt werden darf.
Treten bei jüngeren Patienten venöse und arterielle Thromboembolien ohne anderweitige Risikofaktoren auf, ist die Suche nach Antiphospholipid Antikörpern nebst dem Lupus Antikoagulans Test bereits beim Übersichtslabor mit einzuschliessen. Denn beim hochrisiko Antiphospholipid Syndrom (positiver Lupus Antikoagulans Test, erhöhte Titer Anti-Cardiolipin plus Anti-β2-Glykoprotein-1-Antikörper) ist Rivaroxaban der Behandlung mit Vitamin-K-Antagonisten klar unterlegen (7). Die Behandlung muss mit Vitamin-K-Antagonisten erfolgen.
Abklärung hereditäre Thrombophilie
Zur Abklärung der hereditären Thrombophilien empfehle ich im Setting einer Praxis die funktionelle Bestimmung von Antithrombin und Protein C, die antigenetische Bestimmung des freien Proteins S und die molekulargenetische Suche einer Faktor II (Prothrombinmutation G20210A) und Faktor V (R506Q, Leiden) Mutation. Um pathologische Befunde von Protein C und S interpretieren zu können, ist es unerlässlich zu zeigen, dass zur gleichen Zeit der Bestimmung dieser Inhibitoren die Prothrombinzeit (Quick) normal ist. Ein verminderter Spontanquick könnte ein Hinweis auf einen Vitamin-K-Mangel sein, welcher gleichermassen auch die Vitamin K abhängigen Proteine C und S verringert. Wichtig: Östrogen kann zu einem erworbenen Protein-S-Mangel führen!
Zeitpunkt der Abklärung einer hereditären Thrombophilie
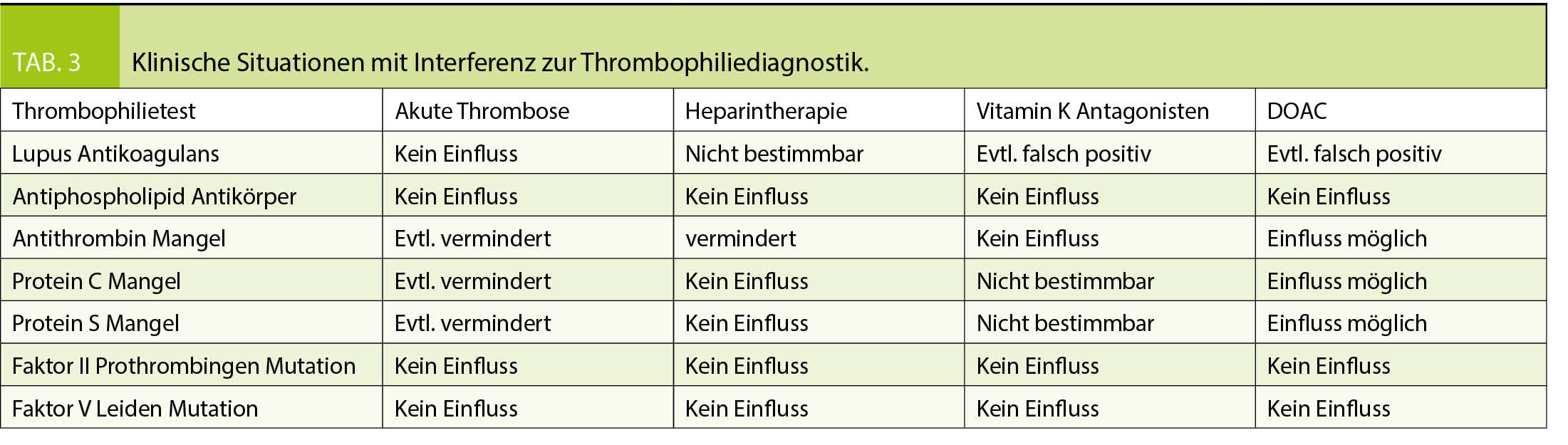
Die Suche einer hereditären Thrombophilie sollte so geplant werden, dass sie unter optimalen Voraussetzungen, d.h. zum richtigen Zeitpunkt erfolgt und eine möglichst abschliessende Beurteilung erlaubt. Diverse Einflüsse können die Thrombophiliediagnostik stören und verfälschen (Tab. 3). Im Idealfall erfolgt die Thrombophilieabklärung 3-4 Wochen nach abgesetzter Antikoagulation. Ist offen, ob eine Indikation für eine Langzeitantikoagulation vorliegt, soll die Abklärung frühestens drei Monate nach dem akuten Ereignis erfolgen. Die Antikoagulation, heute meist ein DOAC, wird 48-72 Std. pausiert und nach erfolgter Blutentnahme fortgesetzt.
Suche nach einer Thrombophilie bei asymptomatischen Patienten
Ob asymptomatische Patienten auf eine Thrombophilie getestet werden sollen bedarf der Abwägung von Vor- und Nachteilen. Kein Nachweis einer Thrombophilie, auch nicht einer solchen mit hohem Risiko für eine Erstthrombose, wie etwa ein Antithrombin Mangel, würde gemäss heutigem Erkenntnisstand prophylaktisch antikoaguliert.
Mögliche Vorteile der Abklärung
Patienten mit bekannter Thrombophilie könnten gezielt instruiert werden die frühen Warnzeichen einer Thromboembolie zu erkennen und beim Auftreten von solchen adäquat zu handeln. Eine gezielte Aufklärung über das erhöhte Risiko bei der Verwendung von Östrogen wird möglich und in Risikosituationen für die Entstehung einer Thromboembolie könnte eine individualisierte Thromboseprophylaxe durchgeführt werden (z.B. niedermolekulares Heparin während der ganzen Schwangerschaft und im Wochenbett bei einem Antithrombin Mangel).
Mögliche Nachteile der Abklärung
Thrombophilien mit hohem Thromboserisiko sind selten und die häufigen Thrombophilien erhöhen das Thromboserisiko wenig (Tab. 2). Thrombophilie Screening Programme würden deshalb teuer. In den USA wurde in einer Kosten-Nutzen-Rechnung geschätzt, dass basierend auf den Zahlen von Todesfällen an Thromboembolien unter kombinierten Kontrazeptiva (3 pro Million Anwenderinnen pro Jahr ohne und 14 pro Million pro Jahr mit heterozygoter Faktor-V-Leiden-Mutation) 92’000 Frauen mit Faktor-V-Leiden-Mutation identifiziert werden müssten, welche anschliessend auf östrogenhaltige Kontrazeptiva verzichten müssten, um einen Todesfall zu verhindern. Um diese Frauen zu identifizieren, müssten über 300 Millionen US$ aufgewendet werden (8). Basierend auf diesen ökonomischen Daten wird davon abgeraten, generell vor der Verschreibung östrogenhaltiger Kontrazeptiva eine hereditäre Thrombophilie zu suchen.
Fazit
Abgesehen vom Nachweis eines Antiphospholipid Syndroms haben Gerinnungsuntersuchungen wenig direkten Einfluss auf den Behandlungspfad einer venösen Thromboembolie. Bei jüngeren Patienten mit unerklärt aufgetretenen venösen und arteriellen Thromboembolien ist rasch nach Antiphospholipid Antikörpern und einem Lupus Antikoagulans zu suchen. Bei Verdacht, dass ein Antiphospholipid Syndrom vorliegt, ist die Antikoagulation überlappend mit niedermolekularem Heparin und einem Vitamin-K-Antagonisten zu starten. Ansonsten hat der Einsatz von Laboruntersuchungen auf der Suche einer Hyperkoagulabilität zurückhaltend und gut überlegt zu erfolgen. Eine angeborene Thrombophilie ist im Kontext einer Thrombose bei jungen Patienten, allen Frauen im gebärfähigen Alter, Personen mit rezidivierenden Thromboembolien und solchen mittleren Alters (45-60 Jahre) mit Kindern und Geschwistern durchzufuhren. Alle hereditären Thrombophilien werden autosomal dominant vererbt. Direkt Verwandte eines lndexpatienten mit Thrombophilie weisen eine solche mit einer Wahrscheinlichkeit von 50% ebenfalls auf. Nur die seltenen hereditären Thrombophilien beeinflussen das Rezidivrisiko (Tab. 2) und somit potentiell die Behandlung. Der Nachweis einer der häufigen hereditären Thrombophilien (Faktor-V-Leiden, Prothrombinmutation) beeinflusst das Rückfallrisiko nicht. Sie erhöhen aber wesentlich das Risiko eine Erstthrombose zu erleiden. Der Nachweis dieser Thrombophilien hilft zwar nicht beim Management des Indexpatienten kann aber helfen, direkt Verwandte mit erhöhtem Risiko für eine Erstthrombose zu identifizieren und diese durch gezielte Beratung und Prophylaxe vor einem solchen Ereignis zu bewahren. Die Suche nach einem Antiphospholipid Syndrom kostet 180, jene nach einer hereditären Thrombophilie rund 430 Franken. Dieser Betrag kann effizient eingesetzt sein, wenn die Befunde helfen, bei direkt Verwandten Untersuchungen zu verhindern oder ganz gezielt durchzuführen. Es ist deshalb wichtig, dass die Befunde dem Patienten mitgeteilt und ausgehändigt werden, so dass er sie mit Angehörigen teilen kann.
Der Autor hat in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
Nur der Nachweis eines Antiphospholipid Syndroms (pos. Lupus Antikoagulans Test, erhöhte Titer von Anti-Cardiolipin und Anti-β2-Glyko-protein 1 IgG und IgM Antikörper) beeinflusst die Behandlung einer Thromboembolie (Behandlung primär mit Vitamin-K-Antagonisten).
Der Nachweis einer hereditären Thrombophilie beeinflusst nur in wenigen Fällen die Behandlung einer Thromboembolie.
Nutzniesser des Nachweises einer hereditären Thrombophilie sind
primär Verwandte ersten Grades, die nach gezielter Untersuchung von einem erhöhten Thromboserisiko erfahren.
Hereditäre Thrombophilien sollten nur bei selektionierten Patienten mit Merkmalen wie: positive Familienanamnese, junges Alter, wiederkehrende Thromboembolien, atypisch lokalisierte Thrombosen
(abdominal, cerebral) gesucht werden.
1. I. S. C. f. W. T. Day, Thrombosis: a major contributor to the global disease burden. Journal of thrombosis and haemostasis : JTH 12, 1580-1590 (2014).
2. A. D. Blann, G. Y. H. Lip, Venous thromboembolism. Bmj 332, 215-219 (2006).
3. F. A. Spencer et al., The Worcester Venous Thromboembolism study: a population-based study of the clinical epidemiology of venous thromboembolism. J Gen Intern Med 21, 722-727 (2006).
4. T. Baglin et al., Clinical guidelines for testing for heritable thrombophilia. British journal of haematology 149, 209-220 (2010).
5. A. N. Nicolaides et al., Prevention and treatment of venous thromboembolism–International Consensus Statement. International angiology : a journal of the International Union of Angiology 32, 111-260 (2013).
6. J. Mateo, A. Oliver, M. Borrell, N. Sala, J. Fontcuberta, Laboratory evaluation and clinical characteristics of 2,132 consecutive unselected patients with venous thromboembolism–results of the Spanish Multicentric Study on Thrombophilia (EMET-Study). Thrombosis and haemostasis 77, 444-451 (1997).
7. V. Pengo et al., Rivaroxaban vs warfarin in high-risk patients with antiphospholipid syndrome. Blood 132, 1365-1371 (2018).
8. M. D. Creinin, R. Lisman, R. C. Strickler, Screening for factor V Leiden mutation before prescribing combination oral contraceptives. Fertil Steril 72, 646-651 (1999).
9. W. M. Lijfering, F. R. Rosendaal, S. C. Cannegieter, Risk factors for venous thrombosis – current understanding from an epidemiological point of view. British journal of haematology 149, 824-833 (2010).
10. S. Z. Goldhaber, Risk factors for venous thromboembolism. Journal of the American College of Cardiology 56, 1-7 (2010).
11. V. De Stefano et al., The Risk of Recurrent Deep Venous Thrombosis among Heterozygous Carriers of Both Factor V Leiden and the G20210A Prothrombin Mutation. New England Journal of Medicine 341, 801-806 (1999).
12. W. K. Ho, G. J. Hankey, D. J. Quinlan, J. W. Eikelboom, Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Archives of internal medicine 166, 729-736 (2006).
13. J. B. Segal et al., Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA : the journal of the American Medical Association 301, 2472-2485 (2009).
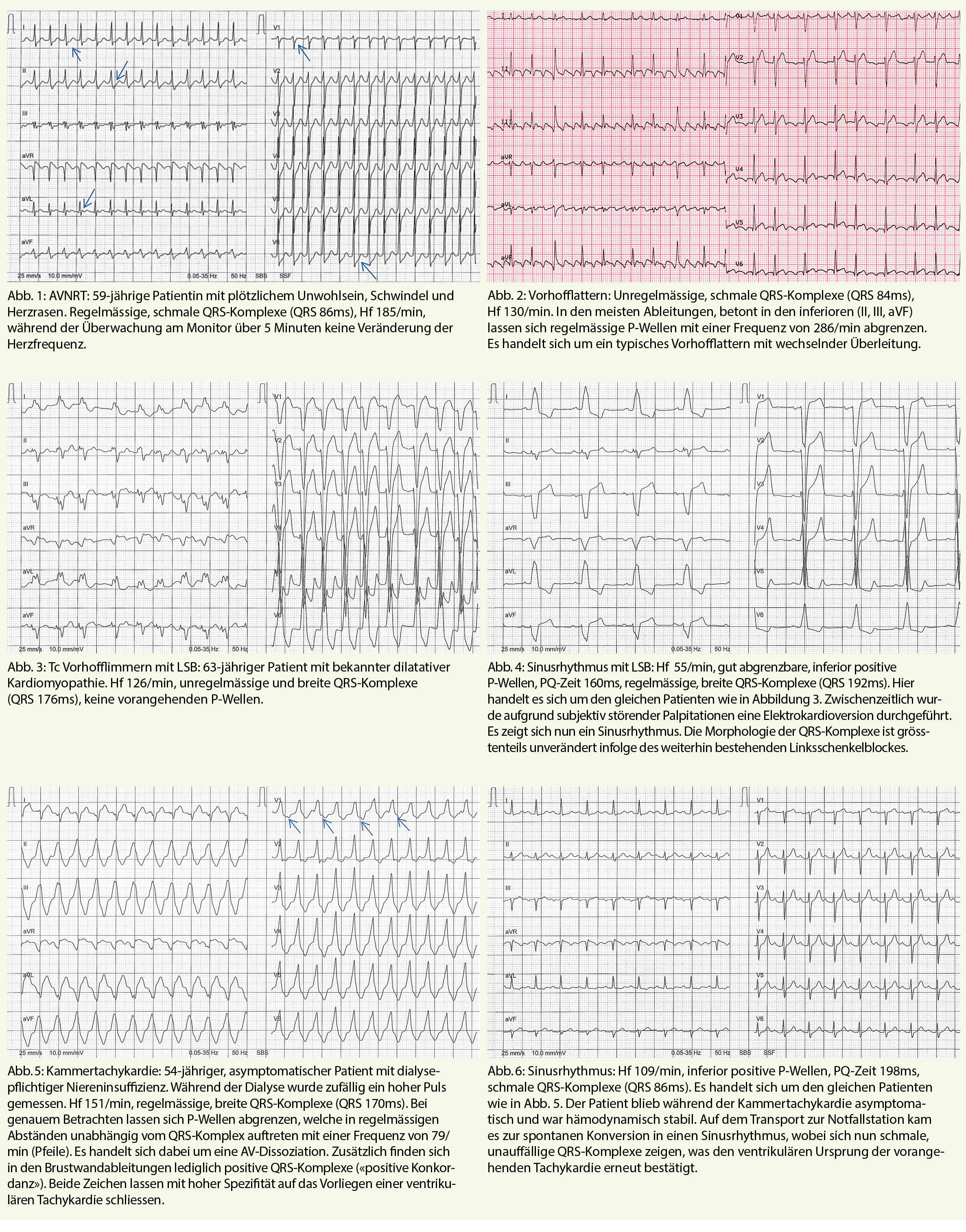
Das Elektrokardiogramm – bereits 1903 durch den holländischen Physiologen Willem Einthoven erfunden – ist aus dem klinischen Alltag nicht mehr weg zu denken. Es ist einfach verfügbar, nicht invasiv und erlaubt eine rasche Diagnose von wichtigen unmittelbar behandlungsbedürftigen kardiologischen Erkrankungen. Es kann einen im klinischen Alltag durchaus vor Herausforderungen stellen, nicht immer ist die Unterscheidung zwischen Artefakt und Pathologie einfach und die Differentialdiagnose mancher EKG-Veränderungen breit. Im Folgenden möchten wir auf die EKG-Charakteristika von tachykarden Herzrhythmusstörungen eingehen.
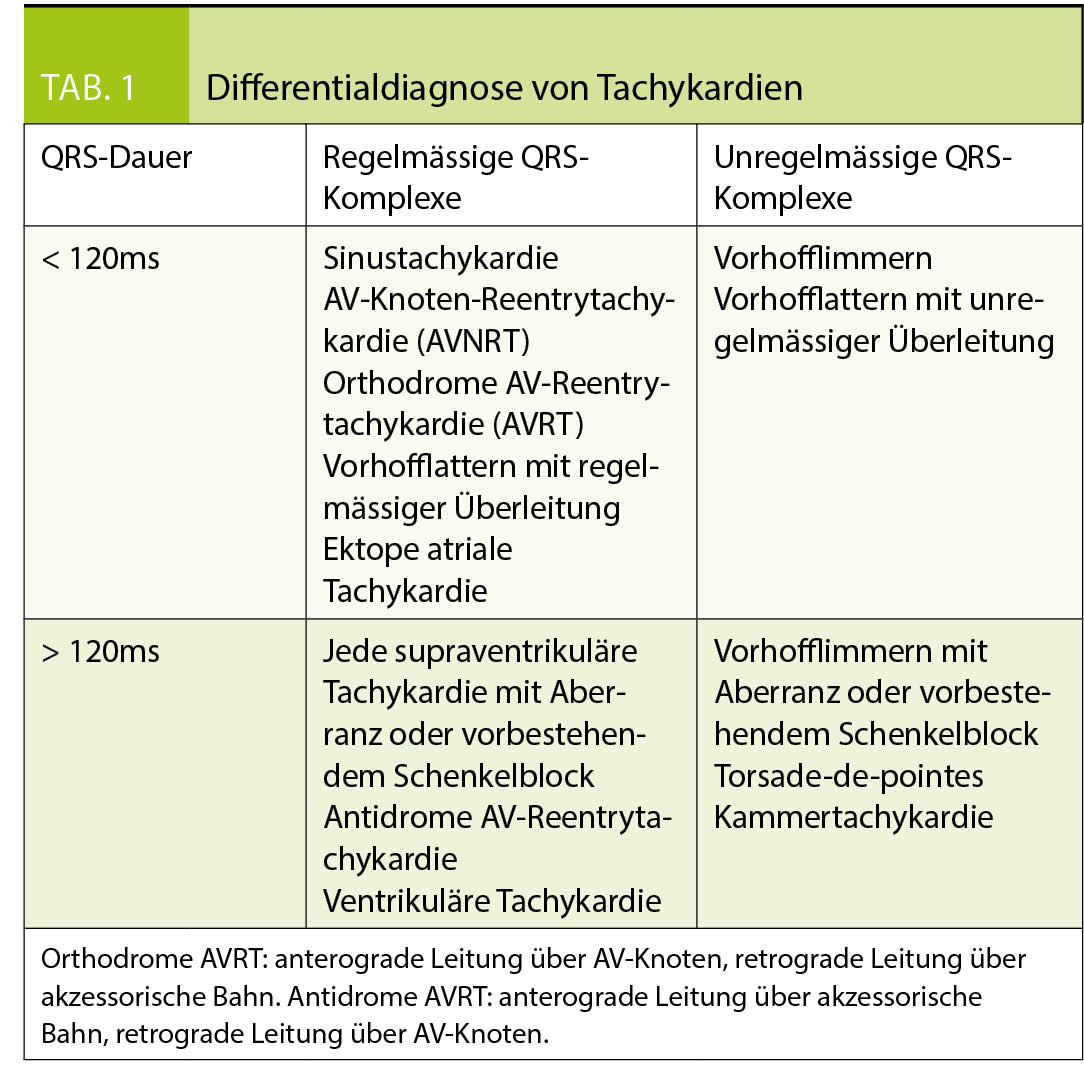
Bei der Analyse von tachykarden Rhythmusstörungen (Herzfrequenz > 100/min) lohnt sich eine Einteilung anhand von Regelmässigkeit und Dauer der QRS-Komplexe (siehe Tabl. 1). Dies lässt eine Eingrenzung der möglichen Differentialdiagnosen zu und erlaubt das Festlegen der initialen Therapiestrategie, auch wenn die definitive Diagnose nicht immer ohne elektrophysiologische Untersuchung gestellt werden kann.
Schmalkomplextachykardien
Findet sich ein schmaler QRS-Komplex bedeutet dies, dass die Erregungsleitung unterhalb des AV-Knotens über das normale Erregungsleitungssystem (His-Bündel, Tawara-Schenkel und Purkinje-Fasern) geleitet wird. Die Tachykardie entsteht somit immer im Vorhof oder AV-Knoten. Am leichtesten lässt sich dabei das Vorhofflimmern von den anderen Arrhythmien abgrenzen, die QRS-Komplexe sind dabei absolut arrhythmisch und P-Wellen fehlen gänzlich.
Bei den Schmalkomplextachykardien mit regelmässigen QRS-Komplexen empfehlen wir einerseits nach sichtbaren P-Wellen zu suchen und andererseits das Frequenzverhalten zu analysieren. Während eine Sinustachykardie und manche ektope atriale Tachykardien frequenzvariabel sind, also zum Beispiel atem- oder belastungsabhängig schneller oder langsamer werden, besteht bei einer Reentry-Tachykardie (AVNRT, AVRT) meist eine relativ starre Herzfrequenz (typischerweise um 140-200/min). Entsprechend berichten die Patienten meist auch über einen abrupten Beginn der Symptomatik. AV-Knoten-Reentry-Tachykardie: In Abbildung 1 zeigen sich beim genauen Hinschauen in den meisten Ableitungen P-Wellen direkt nach dem QRS-Komplex (Pfeile). Es handelt sich um das typische Bild einer AV-Knoten-Reentrytachykardie.
Die P-Wellen entstehen durch die kreisende Erregung im AV-Knoten mit retrograder Erregung der Vorhöfe. Da die mechanische Vorhofkontraktion zeitlich nicht mehr mit der Trikuspidalklappenöffnung synchronisiert ist, können hier Pulsationen in den Halsvenen auftreten («Frog sign»). Vorhofflattern: Beim Vorhofflattern handelt es sich ebenfalls um eine regelmässige Tachykardie im Vorhof (typische Vorhoffrequenz 220-300/min). Jedoch werden diese Vorhoferregungen häufig in wechselnden Abständen auf die Ventrikel weitergeleitet, so dass die QRS-Komplexe unregelmässig auftreten können. Vom Vorhofflimmern kann man dies dadurch differenzieren, dass hier P-Wellen sichtbar sind (typischerweise Sägezahnmuster in den Ableitungen II, III und aVF). Das typische Vorhofflattern entsteht durch einen Reentry-Kreislauf im rechten Vorhof (durch cavotrikuspidalen Isthmus), siehe Abbildung 2. Therapie: Primär sollte bei regelmässigen Schmalkomplextachykardien ein vagales Manöver (Valsalva-Manöver oder Carotismassage) durchgeführt werden. Führt dies nicht zum Erfolg, ist das Medikament der Wahl zur Diagnostik und Therapie Adenosin. Die rasche intravenöse Applikation von 6 mg (bei fehlendem Ansprechen 12 oder maximal 18 mg) führt zu einer Verzögerung oder vollständigen Blockierung der Reizleitung im AV-Knoten über wenige Sekunden. AV-Knoten abhängige Reentrytachykardien (AVNRT oder AVRT) können dadurch häufig terminiert werden. Die anschliessende Therapie der Wahl ist in diesem Fall eine elektrophysiologische Untersuchung mit Ablation der zusätzlichen Leitungsbahn.
Handelt es sich um eine AV-Knoten unabhängige Tachykardie (Vorhofflattern, atriale Tachykardie, ventrikuläre Tachykardie) hat Adenosin meist keinen Einfluss darauf und kann diese nicht terminieren. Aufgrund der kurzzeitigen AV-Blockierung lassen sich aber allfällige P-Wellen besser beurteilen («demaskieren»). Wichtigste Kontraindikation für Adenosin ist neben der hämodynamischen Instabilität ein bekanntes Asthma bronchiale. Gewisse atriale Tachykardien terminieren jedoch unter Adenosin.
Breitkomplextachykardien
Im Gegensatz zu Schmalkomplextachykardien können Breitkomplextachykardien ihren Ursprung im gesamten Herzen haben. Die Wichtigste, meist unmittelbar behandlungsbedürftige und zugleich häufigste Differentialdiagnose ist hier sicherlich die ventrikuläre Tachykardie.
Jedoch kann jede supraventrikuläre Tachykardie mit einem breiten QRS-Komplex einhergehen, wenn das ventrikuläre Myokard neben dem normalen Reizleitungssystem auch über eine akzessorische Bahn erregt wird oder die Reizleitung unterhalb des AV-Knotens zusätzlich beeinträchtigt ist. Letzteres ist der Fall bei vorbestehendem Schenkelblock oder Auftreten von Aberranz (dies bedeutet eine intermittierende Blockierung im Reizleitungssystem bedingt durch unterschiedliche Refraktärzeiten der Leitungsstrukturen also z.B. das Auftreten eines frequenzabhängigen Rechts- oder Linksschenkelblockes).
Hilfreich ist hier, wann immer möglich, der Vergleich mit einem Vor-EKG im supraventrikulären Rhythmus. Besteht eine unveränderte QRS-Morphologie, schliesst dies eine ventrikuläre Tachykardie aus. Des Weiteren wurden diverse Algorithmen entwickelt, welche anhand von vordefinierten EKG-Kriterien in der Differenzierung zwischen ventrikulär und supraventrikulär helfen sollen. Diese vermögen aber alle nicht die Beurteilung im klinischen Kontext zu ersetzen, da keiner eine genügende Sensitivität und Spezifität aufweist. Einer der ersten und in der Praxis häufig verwendeten Algorithmen basiert auf den Brugada-Kriterien (vgl. Tab. 2). Trifft eines der genannten Kriterien zu, erhöht dies die Wahrscheinlichkeit eines ventrikulären Ursprungs der Tachykardien. Vorbestehender Schenkelblock: In Abbildung 3 zeigt sich ein tachykardes Vorhofflimmern bei gleichzeitig bestehendem Linksschenkelblock. Der Linksschenkelblock (LSB) lässt sich im EKG erkennen am breiten QRS-Komplex mit typischerweise überdrehter Linksachse verbunden mit negativen QRS-Komplexen in V1/V2 und positiven QRS-Komplexen V5/V6.
Ein erstmalig dokumentierter LSB ist immer abklärungsbedürftig, um eine zugrundeliegende Erkrankung nicht zu verpassen. Bei unserem 63-jährigen Patienten (Abb. 3 und 4) ist der Schenkelbock auf die dilatative Kardiomyopathie zurück zu führen, was bei dieser Grunderkrankung häufige Folge ist. Weitere häufige Ursachen sind eine koronare Herzkrankheit oder Hypertrophie des linken Ventrikels. Kammertachykardie (Abb 5): Zur Differentialdiagnose dieser regelmässigen Breitkomplextachykardie sind die erwähnten Brugada-Kriterien (siehe Tab. 2) hilfreich.
Abklärung: Die weitere Abklärung inkl. Koronarangiographie zeigte keine strukturelle Kardiopathie oder zugrundeliegende kardiologische Erkrankung. Anhand des EKGs vermuten wir eine benigne Kammertachykardie aus dem linksventrikulären Ausflusstrakt. Therapie: Diese wäre einer elektrophysiologischen Untersuchung und Ablation zugänglich, alternativ kann hier medikamentös ein Klasse 1c-Antiarrhythmikum (z.B. Flecainid) eingesetzt werden.
Jede Tachykardie − selbst die meist gut tolerierten supraventrikulären Reentrytachykardien − ist potentiell kreislaufrelevant. Dafür gefährdet sind insbesondere Patienten mit vorbestehender systolischer oder diastolischer Herzinsuffizienz, da durch die Rhythmusstörung häufig die ventrikuläre Füllung durch eine synchronisierte atriale Kontraktion wegfällt. Die abschliessende Beurteilung des EKGs und Festlegen der Therapie erfordert Erfahrung und häufig auch einen Moment Zeit. Bestehen Zeichen einer hämodynamischen Instabilität, dann ist die Therapie aller Tachykardien die elektrische Kardioversion.
Die Autoren haben in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
Die Eingrenzung der Differentialdiagnosen von Tachykardien gelingt durch die Einteilung in Regelmässigkeit und QRS-Dauer, nach
Möglichkeit soll immer mit einem Vor-EKG verglichen werden.
Bei regelmässigen Schmalkomplextachykardien besteht die erste Massnahme in einem vagalen Manöver, führt dies nicht zum Erfolg,
ist die intravenöse Gabe von Adenosin die Methode der Wahl zur
Diagnostik und Therapie.
Wichtigste – weil potentiell gefährlichste – Differentialdiagnose von Breitkomplextachykardien ist die ventrikuläre Tachykardie. Jedoch kann jede supraventrikuläre Tachykardie mit einem breiten QRS-
Komplex einhergehen, sofern eine Aberranz besteht. Hilfreich zur
Differenzierung sind zum Beispiel die Brugada-Kriterien.
Protonenpumpenhemmer (PPI) gehören zu den meist verschriebenen Medikamenten weltweit. In den letzten Jahren kamen immer mehr Bedenken bezüglich übermässigen Einsatzes und Nebenwirkungen auf. Die meisten der postulierten Nebenwirkungen wurden jedoch lediglich in retrospektiven und Beobachtungsstudien evaluiert und zeigen widersprüchliche und schwache Assoziationen mit einem wesentlichen Risiko. Das Risiko von Nebenwirkungen sollte deshalb kein Grund sein, Ihren Patienten PPI vorzuenthalten. Die richtige Indikation sowie deren Evaluation im Langzeit-Verlauf bleibt das Wichtigste. Nutzen und Risiko werden im Folgenden adaptiert nach (1) diskutiert.
Der Magen ist das einzige Organ, das Säure mit einem pH < 2 produziert. Dies ist sowohl wichtig für das Abtöten von Bakterien, die mit der Nahrung aufgenommen werden, als auch für die Verdauung und die Absorption mehrerer Nahrungsbestandteile wie Proteine, Eisen, Kalzium und Vitamin B12.
Vorteile von PPI in der Langzeitanwendung
PPI blockieren die Säuresekretion während des Tages wirkungsvoll. Die Säureblockade steigt nach peroraler Einnahme von PPI in den ersten 3-5 Tagen graduell an, weil immer nur derjenige Teil der Protonenpumpen blockiert wird, der im aktiven, Säure-sezernierenden Status ist. Um sich an die Protonenpumpen binden zu können, brauchen PPI hochkonzentriertes H2 zur Aktivierung. Diejenigen Protonenpumpen, die gerade nicht aktiv Säure sezernieren, können nicht blockiert werden. Anders als bei den H2-Blockern gibt es deshalb kein Toleranzphänomen, auch nicht in der Langzeitanwendung. V.a. tagsüber verliert die Kraft der Säuresuppression Ihre Wirkung auch mit der Zeit nicht. PPI gehören zu den «Marathonläufern», nicht «Kurzstrecken-Sprintern» (1).
PPI werden fast ausschliesslich über die Leber metabolisiert, nicht über die Niere. Eine Niereninsuffizienz beeinflusst ihre Wirkung deshalb nicht.
Für die Erhaltungstherapie bei gastroösophagealer Refluxerkrankung (GERD) und zur Prophylaxe von gastroduodenalen Ulzera unter NSAR und Aspirin ist eine Langzeitanwendung von PPI notwendig (2, 3). Nicht so bei funktioneller Dyspepsie oder hypersensitivem Ösophagus, wo PPI bei Bedarf angewendet werden können. GERD-Patienten klagen oft über postprandiale Refluxsymptome, weil zur Verdauung der Nahrung Säure produziert wird. Da die Säuresuppression einer einzelnen PPI-Morgendosis für den Tag ausreicht, können PPI 1 x / d effektiv eingesetzt werden, um Reflux oder ösophageale Erosionen / Ulzera zu verhindern. Wichtig ist eine Verabreichung 30-60min vor der Mahlzeit, damit der Wirkstoff im Blut ist, bevor die Säure zur Verdauung der Nahrung produziert wird. Wird die Medikation erst nach dem Essen eingenommen, wenn die gesamte Säure bereits produziert wurde, bleibt die Wirkung praktisch aus. Bei kontinuierlicher Anwendung liegt das Wiederauftreten von GERD-Symptomen während 1 Jahres < 15%, im Gegensatz zu > 50% ohne Erhaltungstherapie (4, 5). Verglichen mit H2-Blockern sind PPI wirksamer. Des Weiteren werden PPI eingesetzt, um die neoplastische Transition eines Barrett-Ösophagus zu Dysplasie / Adenokarzinom längerfristig zu verhindern, obwohl dies nicht ganz klar bewiesen ist (6).
Nachteile von PPI in der Langzeitanwendung
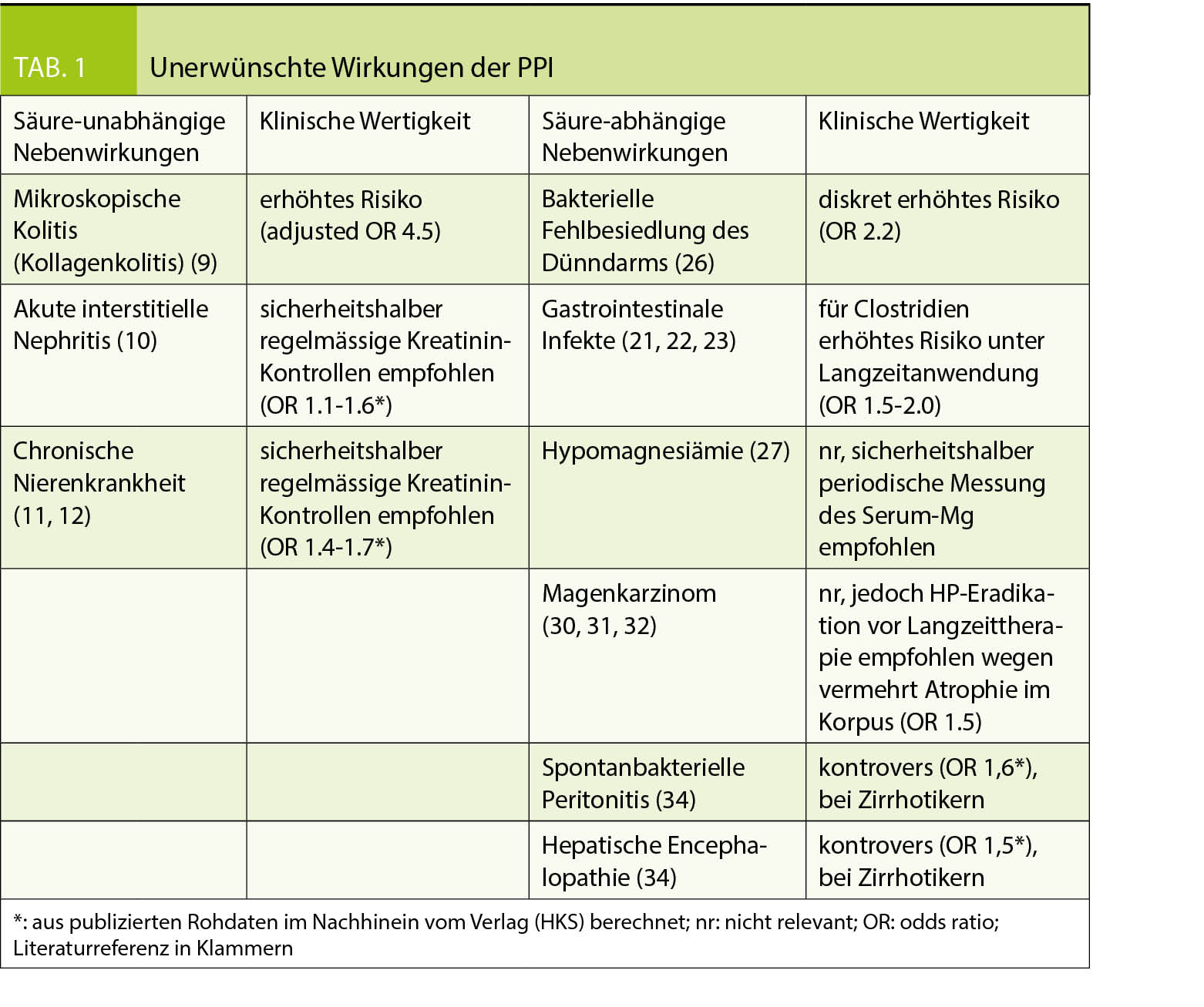
Die Nebenwirkungen der PPI können in 2 Gruppen unterteilt werden: die Säure-unabhängigen und die Säure-abhängigen.
Die meisten Nebenwirkungen der Gruppe, die von der Säureblockade abhängen, treten in der Langzeitanwendung auf. Die von der Säure unabhängigen Nebenwirkungen werden sowohl in der Langzeit- als auch in der Kurzzeitanwendung beobachtet (Tab. 1).
Säure-unabhängige Nebenwirkungen
Mögliche allergische Reaktionen
Anaphylaxie, Panzytopenie, Agranulozytose, Thrombozytopenie, hämolytische Anämie, akuter Leberschaden, Lyell-Syndrom, Stevens-Johnson Syndrom, interstitielle Nephritis und Rhabdomyolyse werden unter PPI selten beschrieben. Dies sind unspezifische allergische Reaktionen, die auch unter anderen Medikamenten auftreten (7, 8). Unerklärte Hautläsionen, Fieber oder generelles Unwohlsein nach Beginn einer PPI-Therapie sollten Ihnen gemeldet werden. Es ist deshalb empfohlen, in den ersten Wochen einer PPI-Therapie wegen möglicher Nebenwirkungen eine Folgekonsultation zu vereinbaren.
Mikroskopische Kolitis (Kollagenkolitis)
Von Patienten unter PPI wird oft über Diarrhoe geklagt. Ein Teil der Fälle kann wahrscheinlich mit einer kollagenen Kolitis erklärt werden, die diagnostiziert wird durch Diarrhoe und histopathologische Veränderungen in der Koloskopie. PPI-Gebrauch ist assoziiert mit erhöhtem Risiko einer kollagenen Kolitis (hazard ratio 4.5) (9). Die PPI-bedingte kollagene Kolitis ist meist selbstlimitierend nach Sistieren der Medikation. Bei Auftreten von Diarrhoe unter neu begonnener PPI-Therapie muss jedoch an eine kollagene Kolitis gedacht werden.
Akute interstitielle Nephritis und chronische Nierenkrankheit
PPI können mit einer interstitiellen Nephritis zusammenhängen, möglicherweise auf Grund einer allergischen Reaktion, wobei der genaue Mechanismus nicht klar ist. In Biopsien wurde von 70% medikamenten-bedingter interstitieller Nephritis berichtet, davon 14% durch PPI (10). Zusätzlich zum akuten Nierenschaden wurde auch von chronischer Nierenkrankheit berichtet, die mit PPI vergesellschaftet sein soll, obwohl die Hazard Ratio bescheiden war (1.1-1.5) und die Resultate nur auf Observationsstudien beruhen (11, 12). Fazit ist, dass Patienten unter PPI sicherheitshalber regelmässig bzgl. Nierenfunktion kontrolliert werden sollten, auch wenn ein Zusammenhang mit PPI nicht klar bewiesen werden konnte.
Medikamenten-Interaktionen
Wie viele andere Medikamente (Diazepam, Phenytoin, Warfarin) werden PPI teilweise durch das Leberenzym CYP2C19 metabolisiert. Die Kapazität dieses Enzyms ist jedoch beschränkt. Deshalb kann die pharmakologische Wirkung anderer Medikamente durch PPI-Gabe beeinflusst werden. Clopidogrel z.B. braucht CYP2C19 zur Aktivierung. Bei Patienten unter Clopidogrel könnten PPI deshalb die anti-thrombotische Wirkung abschwächen und das Risiko für kardiovaskuläre Ereignisse erhöhen. Dies wurde kürzlich postuliert. Zusammenfassend kann aber gesagt werden, dass gemäss aktueller Datenlage (darunter auch die berühmte COGENT-Studie) keine Empfehlung gegen den Gebrauch von PPI bei Patienten mit Clopidogrel besteht (13, 14). Der Evidenzgrad der vorliegenden Studien war nicht adäquat genug, um klinisch relevante Entscheidungen zu fällen.
Demenz
Vor Kurzem wurden 2 retrospektive Publikationen aus deutschen Datenbanken veröffentlicht, in denen bei älteren Patienten unter PPI von einem erhöhten Risiko für Demenz berichtet wurde. Die Hazard Ratio war bescheiden (1.38 und 1.44), es wurde von einem potenziellen cerebralen Schaden durch erhöhte Spiegel von Amyloid-Beta-Peptiden gesprochen. 3 retrospektive Studien in den USA und Europa konnten diese Statistik jedoch nicht bestätigen. Kürzlich widerlegte eine prospektive Populations-Studie diese Aussage sogar eindeutig. PPI sollten nicht vermieden werden wegen Bedenken bezüglich Demenz (15).
Weitere
Kürzlich wurde ein erhöhtes Risiko für zerebrale Ischämie, KHK und sogar verminderte Lebenserwartung im Zusammenhang mit PPI propagiert. Dies waren jedoch retrospektive Studien aus Datenbanken, die für andere Zwecke erstellt worden waren. Es wurde diesbezüglich keine prospektive Untersuchung durchgeführt. Die Verlässlichkeit dieser retrospektiven Studien ist nicht hoch. Nur Odds ratios (OR) von > 2-3 gelten als klinisch relevant. Die OR für diese Krankheiten betrug 1-2 und ist daher klinisch nicht relevant (16).
Säure-abhängige Nebenwirkungen
Pneumonie
Der bakterizide Effekt des Magensaftes ist unter PPI vermindert, weil der Magen-pH erhöht wird. Zusätzlich sollen PPI auch die anti-bakterielle Immunität vermindern, indem sie die lysosomale Enzymaktivität herabsetzen (17). In retrospektiven Studien wurde gezeigt, dass das Pneumonie-Risiko bei GERD-Patienten zwar in den ersten 30 Tagen nach Beginn einer PPI-Therapie, nicht aber im Langzeit-Verlauf anstieg (18). Untersuchungen bei Patienten, die PPI zur Prävention von NSAR-Ulzera bekamen, zeigten kein erhöhtes Risiko für eine Pneumonie (19). Auch eine Meta-Analyse von prospektiven randomisierten Studien ergab kein erhöhtes Pneumonie-Risiko (20).
Gastrointestinale Infekte
Salmonella und Campylobacter sind säure-labile Bakterien und es ist nachvollziehbar, dass sie den Gastrointestinaltrakt allenfalls eher befallen können, wenn die Säureproduktion auf Grund einer PPI-Therapie vermindert ist. Die Datenlage diesbezüglich ist aber kontrovers. Es gibt sowohl Studien, die eine erhöhte Infektanfälligkeit für Salmonella und Campylobacter gezeigt haben, als auch solche, die dies nicht aufzeigen konnten (21, 22).
Die Clostridium difficile-Enteritis ist in westlichen Ländern wegen der ansteigenden Antibiotika-Resistenz ein zunehmendes Problem. Ein erhöhtes Risiko für eine Infektion besteht unter Langzeitanwendung von PPI mit einer OR von 1.5-2.0 (21). Ein Zusammenhang mit komplizierten Krankheitsverläufen oder rezidivierenden C. difficile-Infektionen konnte aber nicht eindeutig festgestellt werden (23).
Neuroendokrine Tumoren des Magens
Nach dem Beginn des weltweiten PPI-Gebrauchs wurden nur vereinzelte Fälle von gastrischen Karzinoid-Tumoren beschrieben. Ein Zusammenhang mit der PPI-Verabreichung ist nicht klar (24). Zusammenfassend kann gesagt werden, dass das Risiko von gastrischen Karzinoid-Tumoren während Langzeitanwendung von PPI klinisch nicht relevant ist.
Mukosale Hypertrophie im Fundus
Eine durch PPI verursachte Hypergastrinämie führt zur mukosalen Hypertrophie im Fundus. Dies ist v.a. bei der Langzeit-Anwendung von PPI der Fall und bei Helicobacter-negativen Patienten. Das abrupte Absetzen einer PPI-Therapie hat deshalb eine Säure-Überproduktion und damit ein Rebound-Phänomen zur Folge (25). Dies macht eine intermittierende PPI-Therapie schwierig.
Veränderungen im Mikrobiom und bakterielle Fehlbesiedlung des Dünndarms
PPI sollen das Mikrobiom verändern und die Zahl der Streptokokken in der Mundhöhle vermehren (26). Die klinische Relevanz einer Mikrobiom-Veränderung durch PPI ist allerdings aktuell nicht klar. Dazu erhöht eine PPI-Therapie die Bakterien-Dichte in Duodenum und Jejunum. Bei > = 100 000 Keimen/ml Dünndarminhalt wird eine bakterielle Fehlbesiedlung des Dünndarms diagnostiziert.
Veränderte Absorption von Mikronährstoffen (Mg, Eisen, Kalzium, Vitamin B12)
Die Hypothese einer Hypomagnesiämie unter PPI basiert auf einer selektiven Malabsorption im Dünndarm, verursacht durch das veränderte Mikro-Milieu bei erhöhtem pH. Die Studienlage ist aber nicht eindeutig (27). Konsequenz davon ist die Empfehlung einer periodischen Messung des Serum-Magnesiums unter Langzeit-Therapie mit PPI.
Einige Mikronährstoffe benötigen die Magensäure für eine effektive Absorption, z.B. Eisen, Kalzium und Vitamin B12. Wenn die Eisenreserven im Körper tief sind, reguliert die duodenale Mukosa automatisch die Eisenabsorption hoch. Deshalb tritt lediglich wegen einer PPI-Therapie selten ein Eisenmangel auf. Bezüglich verminderter Kalzium-Absorption mit daraus resultierender Osteoporose und vermehrten Knochenbrüchen gibt es in verschiedenen Untersuchungen keinen klaren Zusammenhang zur PPI-Gabe (28). Über die Vitamin B12-Absorption unter PPI-Therapie bestehen mehrere Studien, die kontrovers ausfielen und keine klinische Relevanz aufzeigen konnten.
Drüsenkörperzysten
PPI-Therapie bei Helicobacter-negativen Patienten führt gern zur Bildung von multiplen Drüsenkörperzysten. Dies sind kleine, benigne Magenpolypen. Sie verschwinden nach Sistieren der PPI-Therapie wieder. Empfohlen ist eine endoskopische Resektion bei Polypen >1cm, ansonsten haben sie keine klinische Relevanz (29). Bei aussergewöhnlich starkem Auftreten sollte mittels Koloskopie eine familiäre adenomatöse Polyposis (FAP) ausgeschlossen werden, die mit der PPI-Einnahme aber nichts zu tun hat.
Magenkarzinome
Es fehlt die Evidenz, dass eine Langzeitanwendung von PPI zu Magenmukosaatrophie oder Metaplasiebildung führt (30). Patienten mit Helicobacter-Infektion zeigen unter Langzeit-Therapie mit PPI häufiger eine Mukosaatrophie im Korpus als Helicobacter-negative Patienten. Deshalb ist eine Helicobacter-Eradikation vor Langzeit-Therapie mit PPI empfehlenswert. Eine mögliche Folge ist das Fortschreiten der Atrophie und Metaplasie zur Dysplasie. Auch dies kann aber nicht eindeutig belegt werden. Es wurden Magenkarzinome beschrieben, die aber im Zusammenhang mit einer perniziösen Anämie und chronischen Gastritis auftraten, nicht mit einer PPI-Therapie (31, 32).
Kolonkarzinome
Einige Kolonkarzinome haben Gastrinrezeptoren. So könnte eine durch PPI induzierte Hypergastrinämie zu einem erhöhten Kolonkarzinomrisiko führen. Basierend auf mehreren Beobachtungsstudien zeigt sich dafür aber keine Evidenz (33).
Spontanbakterielle Peritonitis und hepatische Encephalopathie
Bei Leberzirrhose und Aszites können wegen erhöhter Permeabilität der intestinalen Mukosa Darmbakterien in den Aszites penetrieren. PPI sollen das Risiko sowohl für eine spontanbakterielle Peritonitis (hazard ratio 1.4-5.0) als auch für das Auftreten einer hepatischen Encephalopathie aus diesem Grund erhöhen. Auch diesbezüglich ist die Datenlage aber kontrovers (34, 35).
Medikamenten-Interaktionen
Die pharmakologische Wirkung von Medikamenten, für welche die Magensäure zur Absorption wichtig ist (z.B. Digoxin), können durch gleichzeitige PPI-Gabe beeinflusst werden. Die Kompatibilität der verschiedenen Medikamente, insbesondere bei Polypharmazie, sollte deshalb auch bei PPI beachtet werden.
Fazit
Am Wichtigsten ist die richtige Indikation vor Verschreibung einer längerfristigen PPI-Therapie. Nebenwirkungen von PPI sind grundsätzlich selten und zeigen eine schlechte Evidenz, treten aber hauptsächlich unter Langzeit-Behandlung auf. Potenzielle Nebenwirkungen sollten die Vorteile der Therapie nicht überragen. Als klinisch relevante Nebenwirkungen können die Kollagenkolitis und bakterielle Fehlbesiedlung des Dünndarms genannt werden. Des Weiteren besteht ein diskret erhöhtes Risiko für gastrointestinale Infekte, v.a. Clostridien. Bei Patienten mit Leberzirrhose sollte wegen möglicher spontanbakterieller Peritonitis und hepatischer Encephalopathie die Indikation für eine PPI-Therapie gut überlegt werden.
Bei der Behandlung von GERD, gastroduodenalen Ulzera oder einer Helicobacter-Eradikation liegen die Vorteile klar auf Seite der PPI-Therapie, während bei funktioneller Dyspepsie oder hypersensitivem Ösophagus die Datenlage für eine erfolgversprechende Therapie limitiert ist. Um lediglich einen geringen therapeutischen Effekt zu erzielen, sollte das Risiko von Nebenwirkungen vermieden werden, auch wenn es klein ist. Wenn der erwartete therapeutische Nutzen hingegen gross ist, kann ein tiefes Nebenwirkungs-Risiko akzeptiert werden.
Dr. med. Mirjam Hiestand
Klinik für Gastroenterologie/Hepatologie
Kantonsspital St. Gallen
Rorschacher Strasse 95
9007 St. Gallen
mirjam.hiestand@kssg.ch
Dr. med. Claudia Krieger-Grübel
Klinik für Gastroenterologie und Hepatologie
HOCH Health Ostschweiz
Kantonsspital St. Gallen
Prof. Dr. med. Jan Borovicka
Klinik für Gastroenterologie/Hepatologie
Kantonsspital St. Gallen
Rorschacher Strasse 95
9007 St. Gallen
Die Autoren/innen geben an, keine Interessens-konflikte im Zusammenhang mit dem vorgelegten Manuskript zu haben.
PPI gehören zu den meistverschriebenen Medikamenten weltweit.
Das Nutzen/Risiko-Profil liegt zu Gunsten der PPI. Nebenwirkungen sind selten und klinisch oft nicht relevant. Die meisten der postulierten Nebenwirkungen wurden in retrospektiven und Beobachtungsstudien evaluiert und zeigen widersprüchliche und schwache Assoziationen mit einem wesentlichen Risiko.
Die beste Evidenz haben die Kollagenkolitis und die bakterielle
Fehlbesiedlung des Dünndarms. Für gastrointestinale Infekte (v.a. Clostridien) besteht ein leicht erhöhtes Risiko. Bei Patienten mit Leberzirrhose sollte wegen möglicher spontanbakterieller Peritonitis und hepatischer Encephalopathie die Indikation für eine PPI-Therapie gut überlegt werden.
Abgewogen werden sollte das Nutzen/Risiko-Profil. Bei geringem therapeutischem Effekt wie funktioneller Dyspepsie sollten PPI möglichst nur bei Bedarf angewendet werden, nicht längerfristig. Bei der Prophylaxe von gastralen Ulzera oder GERD überwiegen jedoch die Vorteile.
1. Yoshikazu Kinoshita, Norihisa Ishimura, and Shunji Ishihara. Advantages and
Disadvantages of Long-term Proton Pump Inhibitor Use. J Neurogastroenterol Motil 2018; 24: 182-196.
2. Labenz J, Armstrong D, Lauritsen K, et al. Esomeprazole 20 mg vs. pantoprazole 20 mg for maintenance therapy of healed erosive oesophagitis: results from the EXPO study. Aliment Pharmacol Ther 2005;22:803-811.
3. Kinoshita Y, Kato M, Fujishiro M, et al. Efficacy and safety of twice- daily rabeprazole maintenance therapy for patients with reflux esophagi- tis refractory to standard once-daily proton pump inhibitor: the Japan- based EXTEND study. J Gastroenterol 2017;28:1-11.
4. Pace F, Annese V, Prada A, et al. Rabeprazole is equivalent to omepra- zole in the treatment of erosive gastro-oesophageal reflux disease. A randomised, double-blind, comparative study of rabeprazole and omeprazole 20 mg in acute treatment of reflux oesophagitis, followed by a maintenance open-label, low-dose therapy with rabeprazole. Dig Liver Dis 2005;37:741-750.
5. Lundell LR, Dent J, Bennett JR, et al. Endoscopic assessment of oe- sophagitis: clinical and functional correlates and further validation of the Los Angeles classification. Gut 1999;45:172-180.
6. Singh S, Garg SK, Singh PP, Iyer PG, El-Serag HB. Acid-suppressive medications and risk of oesophageal adenocarcinoma in patients with Barrett’s oesophagus: a systematic review and meta-analysis. Gut 2014;63:1229-1237.
7. Choi SW, Han JM, Bae YJ, et al. Lessons from two cases of anaphylaxis to proton pump inhibitors. J Clin Pharm Ther 2012;37:614-616.
8. Dury S, Nardi J, Gozalo C, Lebargy F, Deslee G. Agranulocytosis in- duced by
proton pump inhibitors. J Clin Gastroenterol 2012;46:859.
9. Keszthelyi D, Jansen SV, Schouten GA, et al. Proton pump inhibitor use is associated with an increased risk for microscopic colitis: a case-control study. Aliment Pharmacol Ther 2010;32:1124-1128.
10. Muriithi AK, Leung N, Valeri AM, et al. Biopsy-proven acute intersti- tial nephritis, 1993-2011: a case series. Am J Kidney Dis 2014;64:558- 566.
11. Lazarus B, Chen Y, Wilson FP, et al. Proton pump inhibitor use and the risk of chronic kidney disease. JAMA Intern Med 2016;176:238-246.
12. Arora P, Gupta A, Golzy M, Patel N, Carter RL, Jalal K, Lohr JW. Proton pump inhibitors are associated with increased risk of develop- ment of chronic kidney disease. BMC Nephrol 2016;17:112.
13. Gao QP, Sun Y, Sun YX, Wang LF, Fu L. Early use of omeprazole benefits patients with acute myocardial infarction. J Thromb Throm- bolysis 2009;28:282-287.
14. Ren YH, Zhao M, Chen YD, et al. Omeprazole affects clopidogrel ef- ficacy but not ischemic events in patients with acute coronary syndrome undergoing elective percutaneous coronary intervention. Chin Med J 2011;124:856-861.
15. Gray SL, Walker RL, Dublin S, et al. Proton pump inhibitor use and dementia risk: prospective population-based study. J Am Geriatr Soc 2017;66:247-253.
16. Vaezi MF, Yang YX, Howden CW. Complications of proton pump inhibitor therapy. Gastroenterology 2017;153:35-48.
17. Liu W, Baker SS, Trinidad J, et al. Inhibition of lysosomal enzyme activities by proton pump inhibitors. J Gastroenterol 2013;48:1343-1352.
18. Johnstone J, Nerenberg K, Loeb M. Meta-analysis: proton pump inhibitor use and the risk of community-acquired pneumonia. Aliment Pharmacol Ther 2010;31:1165-1177.
19. Filion KB, Chateau D, Targownik LE, et al. Proton pump inhibitors and the risk of hospitalisation for community-acquired pneumonia: rep- licated cohort studies with meta-analysis. Gut 2014;63:552-558.
20. Sultan N, Nazareno J, Gregor J. Association between proton pump in- hibitors and respiratory infections: a systematic review and meta-analysis of clinical trials. Can J Gastroenterol 2008;22:761-766.
21. Bavishi C, Dupont HL. Systematic review: the use of proton pump inhibitors and increased susceptibility to enteric infection. Aliment Phar- macol Ther 2011;34:1269-1281.
22. Brophy S, Jones KH, Rahman MA, et al. Incidence of Campylobacter and
Salmonella infections following first prescription for PPI: a cohort study using
routine data. Am J Gastroenterol 2013;108:1094-1100.
23. Faleck DM, Salmasian H, Furuya EY, Larson EL, Abrams JA, Freed- berg DE.
Proton Pump Inhibitors Do Not Increase Risk for Clostrid- ium difficile Infection in the Intensive Care Unit. Am J Gastroenterol 2016;111:1641-1648.
24. Haga Y, Nakatsura T, Shibata Y, et al. Human gastric carcinoid detected during long-term antiulcer therapy of H2 receptor antagonist and proton pump inhibitor. Dig Dis Sci 1998;43:253-257.
25. Gillen D, Wirz AA, Ardill JE, McColl KE. Rebound hypersecretion after omeprazole and its relation to on-treatment acid suppression and Helicobacter pylori status. Gastroenterology 1999;116:239-247.
26. Jackson MA, Goodrich JK, Maxan ME, et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut 2016;65:749-756.
27. Biyik M, Solak Y, Ucar R, et al. Hypomagnesemia among outpatient long-term proton pump inhibitor users. Am J Ther 2017;24:e52-e55.
28. Hansen KE, Jones AN, Lindstrom MJ, et al. Do proton pump inhibi- tors decrease calcium absorption? J Bone Miner Res 2010;25:2786- 2795.
29. Choudhry U, Boyce HW Jr, Coppola D. Proton pump inhibitor- associated gastric polyps: a retrospective analysis of their frequency, and endoscopic, histologic, and ultrastructural characteristics. Am J Clin Pathol 1998;110:615-621.
30. Song H, Zhu J, Lu D. Long-term proton pump inhibitor (PPI) use and the development of gastric pre-malignant lesions. Cochrane Database Syst Rev. 2014;(12):CDO1O623.
31. Lundell L, Vieth M, Gibson F, Nagy P, Kahrilas PJ. Systematic review: the effects of long-term proton pump inhibitor use on serum gastrin levels and gastric histology. Aliment Pharmacol Ther. 2015;42(6):649-63.
32. Ko Y, Tang J, Sanagapalli S, Kim BS, Leong RW. Safety of proton pump inhibitors and risk of gastric cancers: review of literature and pathophysiological mechanisms. Expert Opin Drug Saf. 2016;15(1):53-63.
33. Robertson DJ, Larsson H, Friis S, Pedersen L, Baron JA, Sørensen HT. Proton pump inhibitor use and risk of colorectal cancer: a popula- tion-based, case-control study. Gastroenterology 2007;133:755-760.
34. Dam G, Vilstrup H, Watson H, Jepsen P. Proton pump inhibitors as a risk factor for hepatic encephalopathy and spontaneous bacterial perito- nitis in patients with cirrhosis with ascites. Hepatology 2016;64:1265- 1272.
35. Kim JH, Lim KS, Min YW, et al. Proton pump inhibitors do not in- crease the risk for recurrent spontaneous bacterial peritonitis in patients with cirrhosis. J Gastroenterol Hepatol 2017;32:1064-1070.
Beim gesunden Erwachsenen liegt die Konzentration des Serum-Bilirubins unter 17 μmol/l, davon weniger als 5% in konjugierter Form. Obgleich Ikterus und Hyperbilirubinämie oft synonym verwendet werden, ist ein Ikterus erst ab einem Serum Bilirubin von mehr als 34 μmol/l, also einem zweifachen des oberen Normwertes, klinisch zu diagnostizieren. Die Gelbverfärbung lässt sich als erstes und am besten am Aussenrand der Konjunktiven und an der oralen Mucosa, insbesondere unter der Zunge erkennen. Die weitere Abklärung eines Ikterus ist kritisch, da es das erste und einzige klinische Zeichen einer relevanten Lebererkrankung sein kann (1).
Eine Hyperbilirubinämie lässt sich nach dem überwiegenden Gallenpigment in zwei Kategorien einteilen:
1. Erhöhung des unkonjugierten (indirekten) Serum-Bilirubins z.B. durch vermehrte Bildung von Bilirubin, gestörte Aufnahme von Bilirubin in die Leber oder gestörte Bilirubinkonjugation.
2. Erhöhung des unkonjugierten wie auch des konjugierten Bilirubins aufgrund einer hepatozellulären Erkrankung, Störung der kanalikulären Exkretion, gestörte Wiederaufnahme von konjugiertem Bilirubin oder einer Gallenwegsobstruktion (1).
Indirekte Hyperbilirubinämie
A. Gesteigerte Bildung von Bilirubin
a. Extravaskuläre Hämolyse – im Rahmen der meisten hämolytischen Erkrankungen kommt es zu einem vermehrten Abbau von Erythrozyten durch phagozytierende Zellen in Milz, Knochenmark und Leber.
b. Extravasation von Erythrozyten – Beim Austritt von Erythrozyten in die Pleura- oder Peritonealhöhle oder ins Gewebe kommt es zur Phagozytose durch Gewebsmakrophagen und zum vermehrten Anfall von Bilirubin.
c. Intravaskuläre Hämolyse – Hierbei wird Bilirubin vor allem in Leber und Nieren gebildet und intravasal an Haptoglobin gebunden. Haptoglobin kann bei ausgeprägter Hämolyse depletiert werden.
d. Dyserythropoese – gestörter Einbau von Bilirubin in die Erythrozyten, wie unter anderem bei der megaloblastischen und sideroblastischen Anämie, schwerer Eisenmangelanämie oder Bleivergiftungen (2).
e. Hämolyse bei körperlichem Stress – hierbei kann es zu einem bis zu 10fachen Anstieg der Bilirubinproduktion kommen. Bei Patienten mit normaler Leberfunktion ist allerdings die Kapazität für die Konjugation weit höher als selbst ein massiver Anfall von unkonjugiertem Bilirubin, so dass die kanalikuläre Exkretion zum geschwindigkeitsbestimmenden Schritt wird (3). Konjugiertes Bilirubin wird dann renal eliminiert und die Serumspiegel steigen nicht über 68 μmol/l. Beim Leberkranken hingegen kann es zu erheblichen Anstiegen des Serum Bilirubins kommen. Bei diesen Patienten kommt es zu einem gemischten Anstieg von unkonjugiertem und konjugiertem Bilirubin.
f. Im Gegensatz dazu findet sich bei Patienten mit einer erblichen Störung der Konjugation, wie z.B. dem Morbus Meulengracht, eine isolierte unkonjugierte Hyperbilirubinämie, da der geschwindigkeitsbestimmende Schritt der Elimination hier die Konjugation ist (4).
g. Von besonderer Bedeutung ist der Bilirubin Spiegel für die Diagnose des akuten Leberversagens bei Morbus Wilson. In dieser Situation findet sich eine normale oder ungewöhnlich niedrige alkalische Phosphatase (AP) wohingegen aufgrund der pathognomonischen Hämolyse die Bilirubinwerte hoch sind. Beim erwachsenen Patienten mit akutem Leberversagen liegt die Sensitivität einer AP (IU/ml)/Bilirubin (mg/dl) Ratio von < 4 bei 94, die Spezifität sogar bei 96% (5).
B. Verminderter Abbau von Bilirubin
a. Gestörte hepatische Bilirubinaufnahme – Sowohl eine Störung des Bilirubintransports in die Leber wie auch eine gestörte Aufnahme in den Hepatozyten können zu erhöhten Bilirubinspiegeln führen. Ersteres kann durch eine Herzinsuffizienz oder portosystemische Shunts bedingt sein, letzteres medikamentös (z.B. Rifampicin, Probenecid) sowie in manchen Fällen des
Morbus Meulengracht.
b. Gestörte Bilirubinkonjugation – eine reduzierte Glucuronidierung infolge verminderter oder fehlender UDP-Glucuronosyltransferase-Aktivität ist Merkmal einiger erworbener und erblicher Erkrankungen. Hierzu zählen Crigler-Najjar Syndrom Typ I und Typ II sowie der bereits erwähnte Morbus Meulengracht. Hyperthyroidismus und Ethinylestradiol-haltige Kontrazeptiva wie auch einige Antibiotika (Gentamicin) können die Glucuronidierung inhibieren (6). Auch fortgeschrittene Leberschäden gehen oft mit einer reduzierten Glucuronidierung einher.
Direkte Hyperbilirubinämie
Bei den erworbenen Erkrankungen, die zu einer direkten Hyperbilirubinämie führen, lassen sich einige grundlegende Pathomechanismen unterscheiden: Obstruktion der Gallen-wege mit extrahepatischer Cholestase, intrahepatische Cholestase und hepatozelluläre Schädigungen.
A. Gallenwegsobstruktion – es akkumulieren sowohl unkonjugiertes wie auch konjugiertes Bilirubin innerhalb der Hepatozyten, hierbei kann es auch zur Dekonjugation kommen und erneut dekonjugiertes Bilirubin gebildet werden (7). Ikterus aufgrund einer Gallenwegsobstruktion beim Erwachsenen hat zahlreiche Differentialdiagnosen: Gallensteinleiden mit der Sonderform des Mirizzi-Syndrom, bei dem die durch einen Zystikusstein gestaute Gallenblase den Hauptgallengang komprimiert. Daneben Tumoren mit intra- oder extraluminaler Obstruktion, Primär Sklerosierende Cholangitis, die intra- wie extrahepatische Gallenwege betreffen kann, parasitärer Befall mit Ascaris oder Leberegeln (Chlonorchis und Fasciola), Lymphome, AIDS Cholangiopathie, die durch Cryptosporidien, CMV oder aber HIV selbst verursacht wird (8), akute und chronische Pankreatitis sowie postinterventionelle Strikturen.
B. Intrahepatische Cholestase – hierbei findet sich ein Ikterus sowie eine erhöhte Serum AP so dass das Bild einer Gallengangsobstruktion besteht, die Gallenwege aber frei sind. Wichtige Differentialdiagnosen sind: Virale Hepatitiden, die sich mit cholestatischem Bild und starkem Pruritus manifestieren können. Alkoholische Steatohepatitis (ASH) die sich mit Cholestase, Fieber und Leukozytose präsentieren kann (9). Nichtalkoholische Steatohepatitis (NASH) zeigt klinisch wie auch histologisch Ähnlichkeiten mit der ASH. Primär Biliäre Cholangitis zeigt typischer Weise ein cholangitisches Bild, allerdings finden sich auch hepatozelluläre Schäden. Pharmaka und Toxine können dosisabhängig (anabole Steroide, Ethinylestradiol) oder seltener im Sinne eines allergischen Geschehens «idiosynkratisch» zu einer Cholestase führen. Daraus ergibt sich, dass eine sorgfältige Medikamenten- und Substanzanalyse bei jeder Cholestase unerlässlich ist. Besonders erwähnt werden sollen hier unkontrollierte Phytotherapeutika und «pflanzlichen Produkte», die Ursache für unklare Cholestase und Leberschäden sein können (10).
Sepsis und septisches Kreislaufversagen können ebenfalls zu einem cholestatischen Bild führen. Daneben kann Cholestase als paraneoplastisches Syndrom (Stauffer Syndrom) insbesondere bei Nierenzellkarzinomen, gynäkologischen Malignomen und Prostatakarzinomen beobachtet werden (11). Infiltrationen des Leberparenchyms durch pathologische Prozesse wie Amyloidose, Lymphome oder Tuberkulose können ebenfalls ursächlich sein. Bei den erblichen Erkrankungen mit intrahepatischer Cholestase und erhöhtem konjugiertem Bilirubin sind das Dubin-Johnson Syndrom, das Rotor Syndrom, die progressive familiäre intrahepatische Cholestase (PFIC), die benigne rekurrente intrahepatische Cholestase (BRIC) und die low phospholipid assoziierte Cholelithiasis (LPAC) zu nennen. Bereits in der Neugeborenenperiode manifestieren sich Alagille Syndrom, Cystische Fibrose und manche angeborenen Störungen des Kohlenhydrat-, Fett- oder Gallenmetabolismus mit konjugierter Hyperbilirubinämie. Totale parenterale Ernährung kann Lebersteatose und Cholestase verursachen, wobei vorbestehende Leberschäden ein Risikofaktor sind (12).
Bei der Sichelzellanämie kann es im Rahmen der Hepatischen Krise zu dramatischen Erhöhungen des Bilirubins und der Gallensäuren kommen (13).
Eine weitere Entität stellt die Intrahepatische Schwangerschafts-cholestase (ICP) dar. Meist steht Pruritus als Anfangssymptom im Vordergrund, im weiteren Verlauf kann Ikterus folgen. Hier ist das erhöhte Schwangerschaftsrisiko zu bedenken das mit hohen Gallensäurespiegeln einhergeht (14).
C. Hepatozelluläre Schädigung – Bei einer primären hepatozellulären Schädigung stehen in der Laborkontrolle die erhöhten Transaminasen im Vordergrund, die aber von erhöhtem Bilirubin und Gallensäuren begleitet werden können.
Zusammenfassend
kann dem klinischen Bild des Ikterus und dem laborchemischen Befund der Hyperbilirubinämie eine Vielzahl von Erkrankungen zugrunde liegen. Hilfreich ist die Differenzierung des Serum- Bilirubins und damit eine erste diagnostische Einordnung. Eine erschöpfende Abklärung des Ikterus ist angesichts der möglichen gravierenden Grunderkrankungen unbedingt erforderlich.
Dr. med. Joachim Carl Philipp Mertens
Klinik für Gastroenterologie und Hepatologie
Universitätsspital
8091 Zürich
joachim.mertens@usz.ch
Der Autor hat in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
Die Ursachen des Ikterus sind vielfältig und können hepatischen wie nicht-hepatischen Ursprungs sein
Die Unterscheidung von indirekter und direkter Hyperbilirubinämie ist für die Differenzialdiagnose wichtig
Eine relevante Lebererkrankung sollte bei jedem Ikterus ausgeschlossen werden.
1. Reisman Y, Gips CH, Lavelle SM, Wilson JH. Clinical presentation of (subclinical) jaundice–the Euricterus project in The Netherlands. United Dutch Hospitals and Euricterus Project Management Group. Hepatogastroenterology 1996; 43:1190-5.
2. Robinson S, Vanier T, Desforges JF, Schmid R. Jaundice in thalassemia minor: a consequence of “ineffective erythropoiesis”. N Engl J Med 1962; 267:523-9.
3. Berk PD, Wolkoff AW, Berlin NI. Inborn errors of bilirubin metabolism. Med Clin North Am 1975; 59:803-16.
4. Jansen PL, Roskams T. Why are patients with liver disease jaundiced? ATP-
binding cassette transporter expression in human liver disease. J Hepatol 2001; 35:811-3.
5. Korman JD, Volenberg I, Balko J, Webster J, Schiodt FV, Squires RH, Jr., Fontana RJ, Lee WM, Schilsky ML, Pediatric, Adult Acute Liver Failure Study G. Screening for Wilson disease in acute liver failure: a comparison of currently available
diagnostic tests. Hepatology 2008; 48:1167-74.
6. Chowdhury JR, Chowdhury NR, Moscioni AD, Tukey R, Tephly T, Arias IM. Differential regulation by triiodothyronine of substrate-specific uridinediphosphoglucuronate glucuronosyl transferases in rat liver. Biochim Biophys Acta 1983; 761:58-65.
7. Rius M, Nies AT, Hummel-Eisenbeiss J, Jedlitschky G, Keppler D. Cotransport of reduced glutathione with bile salts by MRP4 (ABCC4) localized to the basolateral hepatocyte membrane. Hepatology 2003; 38:374-84.
8. McCarty M, Choudhri AH, Helbert M, Crofton ME. Radiological features of AIDS related cholangitis. Clin Radiol 1989; 40:582-5.
9. Mendenhall CL, Anderson S, Weesner RE, Goldberg SJ, Crolic KA. Protein-calorie malnutrition associated with alcoholic hepatitis. Veterans Administration Cooperative Study Group on Alcoholic Hepatitis. Am J Med 1984; 76:211-22.
10. Tandon BN, Joshi YK, Sud R, Koshy A, Jain SK, Tandon HD. Follow-up of
survivors of epidemic veno-occlusive disease in India. Lancet 1984; 1:730.
11. Dourakis SP, Sinani C, Deutsch M, Dimitriadou E, Hadziyannis SJ. Cholestatic jaundice as a paraneoplastic manifestation of renal cell carcinoma. Eur J Gastroenterol Hepatol 1997; 9:311-4.
12. Allardyce DB. Cholestasis caused by lipid emulsions. Surg Gynecol Obstet 1982; 154:641-7.
13. Betrosian A, Balla M, Kafiri G, Palamarou C, Sevastos N. Reversal of liver failure in sickle cell vaso-occlusive crisis. Am J Med Sci 1996; 311:292-5.
14. Dixon PH, Weerasekera N, Linton KJ, Donaldson O, Chambers J, Egginton E, Weaver J, Nelson-Piercy C, de Swiet M, Warnes G, Elias E, Higgins CF, Johnston DG, McCarthy MI, Williamson C. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Hum Mol Genet 2000; 9:1209-17.