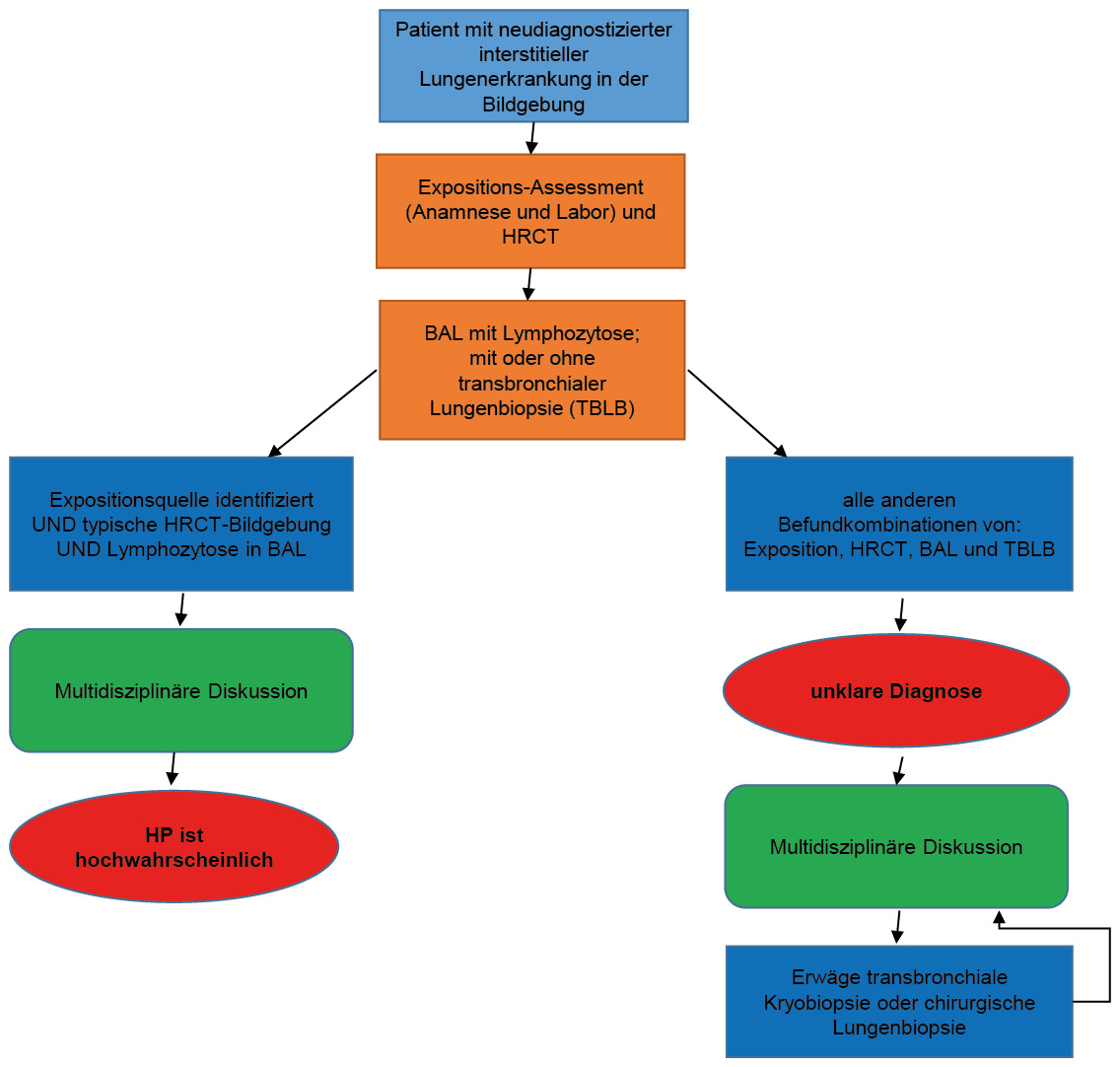
Kardiovaskuläre Erkrankungen (cardiovascular diseases, CVD) stellen weltweit und in der Schweiz ein erhebliches Gesundheitsproblem dar. Trotz präventiver Massnahmen und Fortschritten in der Behandlung führen kardiovaskuläre Erkrankungen in der Schweiz immer noch zu einer beträchtlichen Zahl von Hospitalisierungen (133 000 in 2021) und sind für fast 1/3 aller Todesfälle (19 600 in 2021) verantwortlich. Die Notfallversorgung akuter kardiovaskulärer Ereignisse weist heute in der Schweiz im Vergleich zum Ausland einen sehr hohen Standard auf. Doch es besteht eine grosse Diskrepanz aus Evidenz und täglicher Praxis (Evidence-Performance-Gap) in der kardiovaskulären Risikofaktorkontrolle, denn ein grosser Prozentsatz der Patient/-innen erreicht die jeweiligen Ziele der Leitlinienempfehlungen nicht: 55 % der Hypertoniker, 81 % der Patient/-innen mit erhöhtem LDL-Cholesterin und 44 % der Diabetiker. Darüber hinaus rauchen in der Schweiz aktuell 21 %, 42 % gelten als adipös und 24 % der Menschen weisen einen Bewegungsmangel auf.
1. Die globale und schweizweite Krankheitslast (Burden of Disease) kardiovaskulärer Erkrankungen
1.1 Todesfälle und Hospitalisierungen
Kardiovaskuläre Erkrankungen (cardiovascular diseases, CVD) sind gemäss World Health Organization (WHO) global für 31 % (17,9 Millionen) aller Todesfälle verantwortlich (1, 2). Da in den Industriestaaten Infektionskrankheiten und Traumata als Todesursache seltener vorkommen als in weniger entwickelten Ländern, ist der relative Anteil von kardiovaskulären Erkrankungen an den Todesfällen in Europa und den USA sogar noch deutlich höher. Zudem ist die Bevölkerung in den Industriestaaten älter, und es herrschen andere Lebensstile und andere Ernährungsgewohnheiten. Die WHO und die Krankheitsstatistik der Europäischen Gesellschaft für Kardiologie (ESC) gehen daher davon aus, dass in Europa bis zu 45 % (3,9 Millionen) und damit fast die Hälfte aller Todesfälle auf kardiovaskuläre Erkrankungen zurückgehen (2).
In der Schweiz sind kardiovaskuläre Erkrankungen für fast 1/3 aller Todesfälle verantwortlich (1). Alarmierend ist insbesondere die Tatsache, dass gemäss dem Schweizerischen Gesundheitsobservatorium (Obsan) ein kontinuierlicher Anstieg der kardiovaskulären Erkrankungen zu verzeichnen ist. Während 2007 noch 13,9 % an einer kardiovaskulären Erkrankung litten, stieg der Prozentsatz 2012 bereits auf 17,7 % (letztverfügbare Daten bei Obsan), wobei hier auch eine Zunahme der älteren Bevölkerung und eine bessere Diagnostik eine Rolle spielen könnten (3).
Im Gegensatz zur steigenden Prävalenz der kardiovaskulären Erkrankungen respektive kardiovaskulärer Risikofaktoren ist die Mortalität aufgrund von kardiovaskulären Erkrankungen in den Industriestaaten in den letzten Jahren rückläufig, was sowohl auf eine bessere Prävention (primär wie sekundär) als auch auf eine bessere (Akut-)Therapie zurückgeführt wird (4, 5). Trotzdem gab es im Jahr 2021 rund 19 600 Todesfälle und 133 000 Hospitalisierungen durch kardiovaskuläre Erkrankungen in der Schweiz, wobei 33 097 Hospitalisierungen auf ischämische Herzerkrankungen und 18 516 Hospitalisierungen auf einen Schlaganfall zurückzuführen sind. Allein diese beiden Erkrankungen waren zudem für 6 311 (ischämische Herzkrankheiten) respektive 2 663 (Schlaganfälle) Todesfälle verantwortlich (6).
1.2 Epidemiologie und Versorgung der kardiovaskulären Erkrankungen
Epidemiologisch gibt es zwei gegenläufige Trends: einerseits die Alterung der Bevölkerung und somit einen immer grösseren Bevölkerungsanteil mit kardiovaskulären Risikofaktoren oder kardiovaskulären Erkrankungen und andererseits eine verbesserte medizinische Versorgung. Insbesondere die Notfallversorgung akuter kardiovaskulärer Ereignisse weist heute in der Schweiz im Vergleich zum Ausland einen sehr hohen Standard auf. Dies gilt für die Diagnostik wie auch die Akuttherapie im Spital. In nahezu allen Hausarztpraxen existiert ein Point-of-Care-Labor mit der Möglichkeit für Troponin-Schnelltests, eine rasche Notfalllogistik für den Transport ins Spital und eine flächendeckende Infrastruktur mit Plätzen für eine Herzkatheterintervention. Somit ist eine zeitnahe Intervention innert des kritischen Zeitfensters in der ganzen Schweiz sichergestellt. Daher bietet die Primär- und Sekundärprävention das grösste Potenzial zur weiteren Reduzierung der kardiovaskulären Morbidität und Mortalität. Dies gilt insbesondere, da ein grosser Teil der Patient/-innen die vorgegebenen Ziele der ESC-Leitlinien nicht erreichen, obwohl beispielsweise das Non-HDL-Cholesterin in den ESC-Mitgliedsstaaten sukzessive sinkt (2).
1.3 Einfluss auf Lebensquantität und Lebensqualität
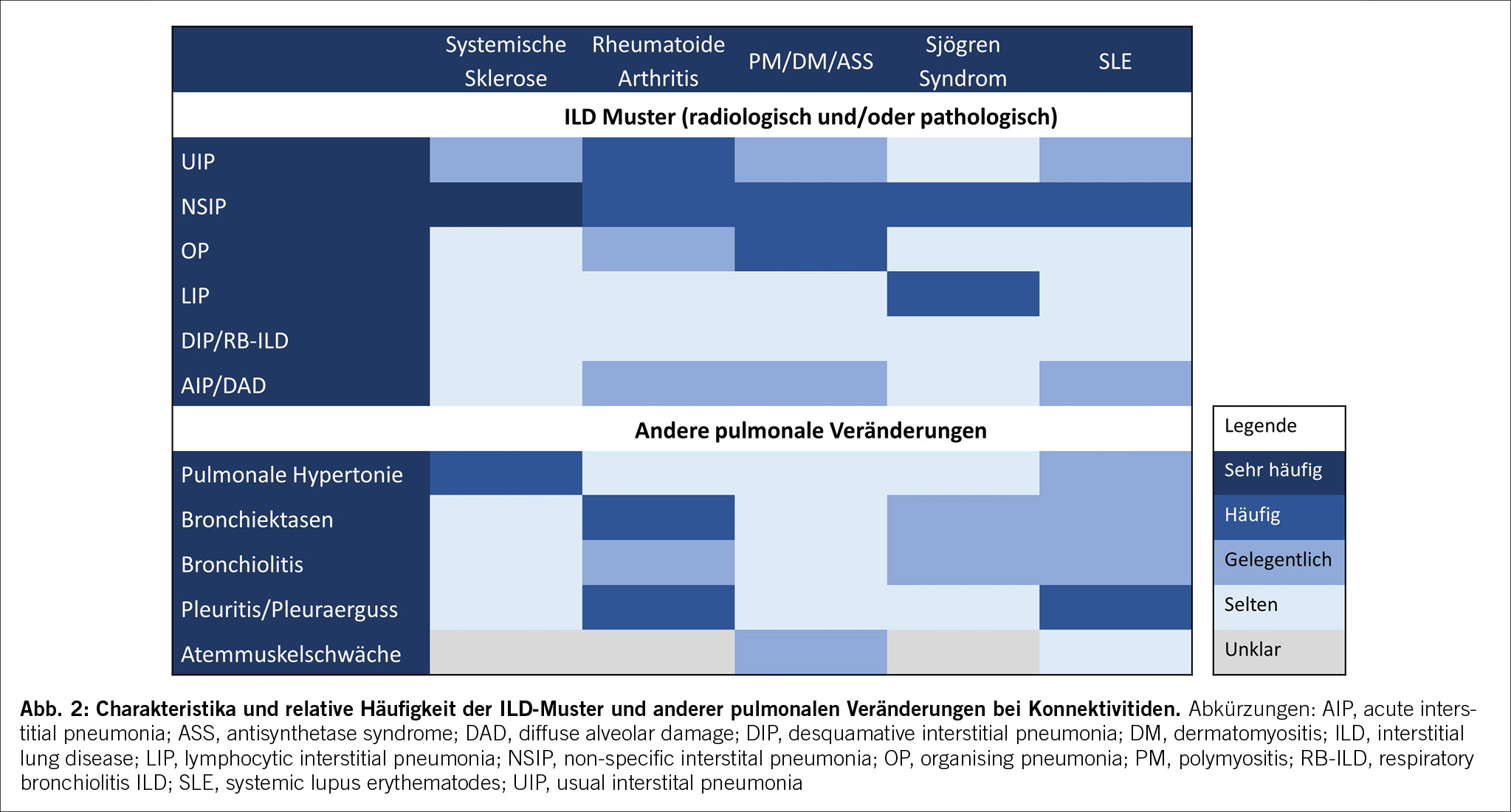
Neben der Verhinderung von Todesfällen, die sich in der Masszahl der gewonnenen Lebensjahre (life years gained, LYG) ausdrücken lässt, sind kardiovaskuläre Erkrankungen auch mit einer erheblichen Morbiditätslast, also einer Einschränkung der Lebensqualität, verbunden. Die disability- (oder disease-) adjusted life years (DALYs) quantifizieren den negativen Einfluss einer Erkrankung, indem sie sowohl die Summe der verlorenen Lebensjahre (life years lost, YLL) wie auch die Lebensjahre mit einer gesundheitlichen Beeinträchtigung (years lived with disability, YLD), etwa infolge eines Herzinfarktes oder eines Schlaganfalls, aufaddieren. Abbildung 1 zeigt deutlich, wie insbesondere ischämische Herzerkrankungen mit einem Verlust an Lebensjahren assoziiert sind.
Neben der massiven Beeinträchtigung der Lebensquantität und Lebensqualität beispielsweise durch Herzinfarkte oder Schlaganfälle weisen kardiovaskuläre Erkrankungen auch eine erhebliche ökonomische Komponente auf. Einerseits direkt durch die Kosten, die aus der (Akut-)Versorgung resultieren, andererseits durch die indirekten Kosten, weil Erkrankte nicht erwerbstätig sein können und somit zum Produktivitätsverlust der Gesellschaft führen. Weiter können auch vorzeitig Verstorbene keinen ökonomischen Beitrag an die Gesellschaft leisten und mindern somit das Bruttoinlandsprodukt.
2. Die bedeutendsten kardiovaskulären Risikofaktoren und die Lücke in der Zielwerterreichung
Dieses Papier untersucht, wie viele DALYs für kardiovaskuläre Risikopatient/-innen durch eine evidenzbasierte Behandlung gewonnen werden können. Die evidenzbasierte Behandlung orientiert sich an den vorhandenen Leitlinien und deren klinischen Zielwerten. Zugleich soll dieses Papier aufzeigen, welche Kosten und welche sozioökonomische Last damit vermieden werden könnten.
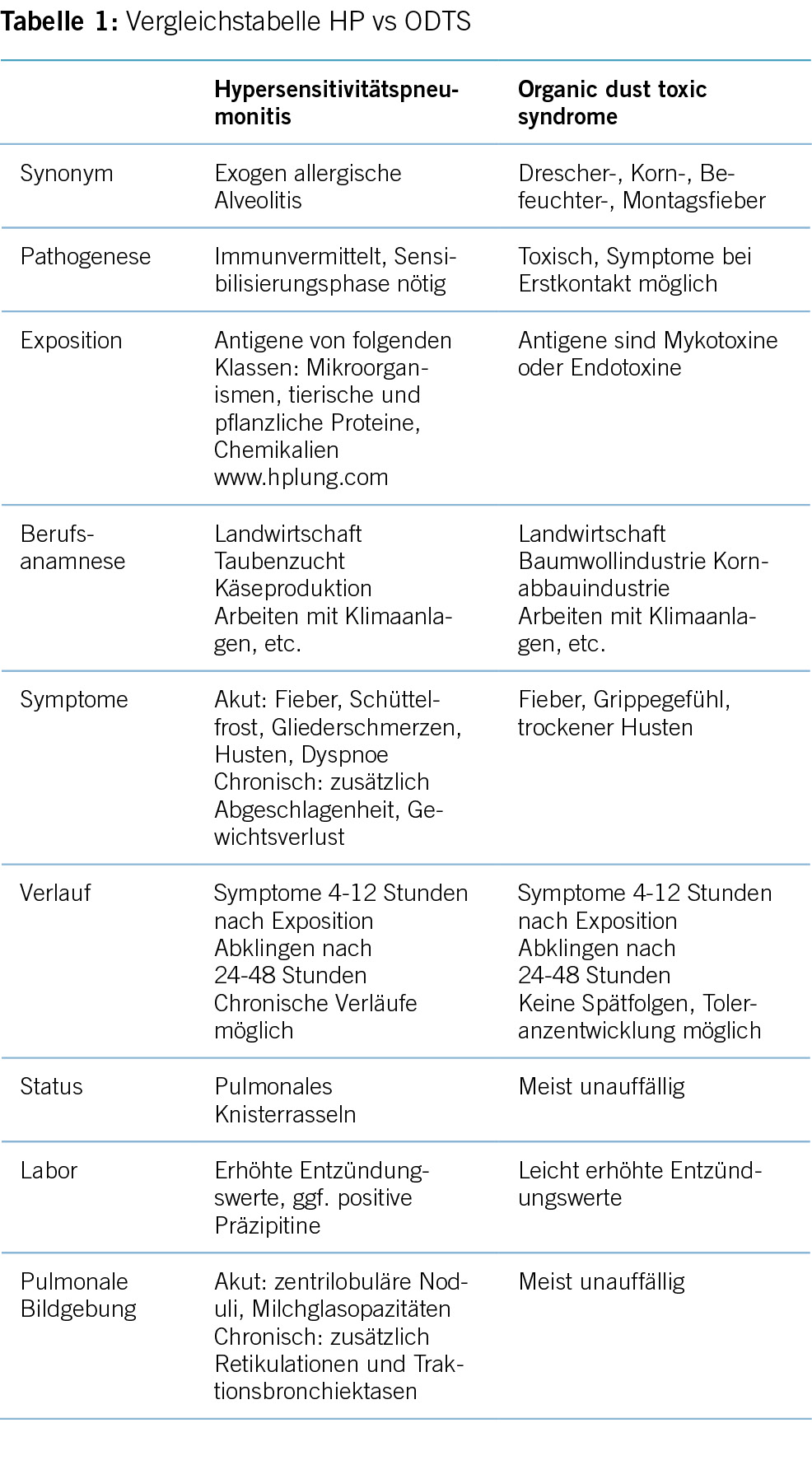
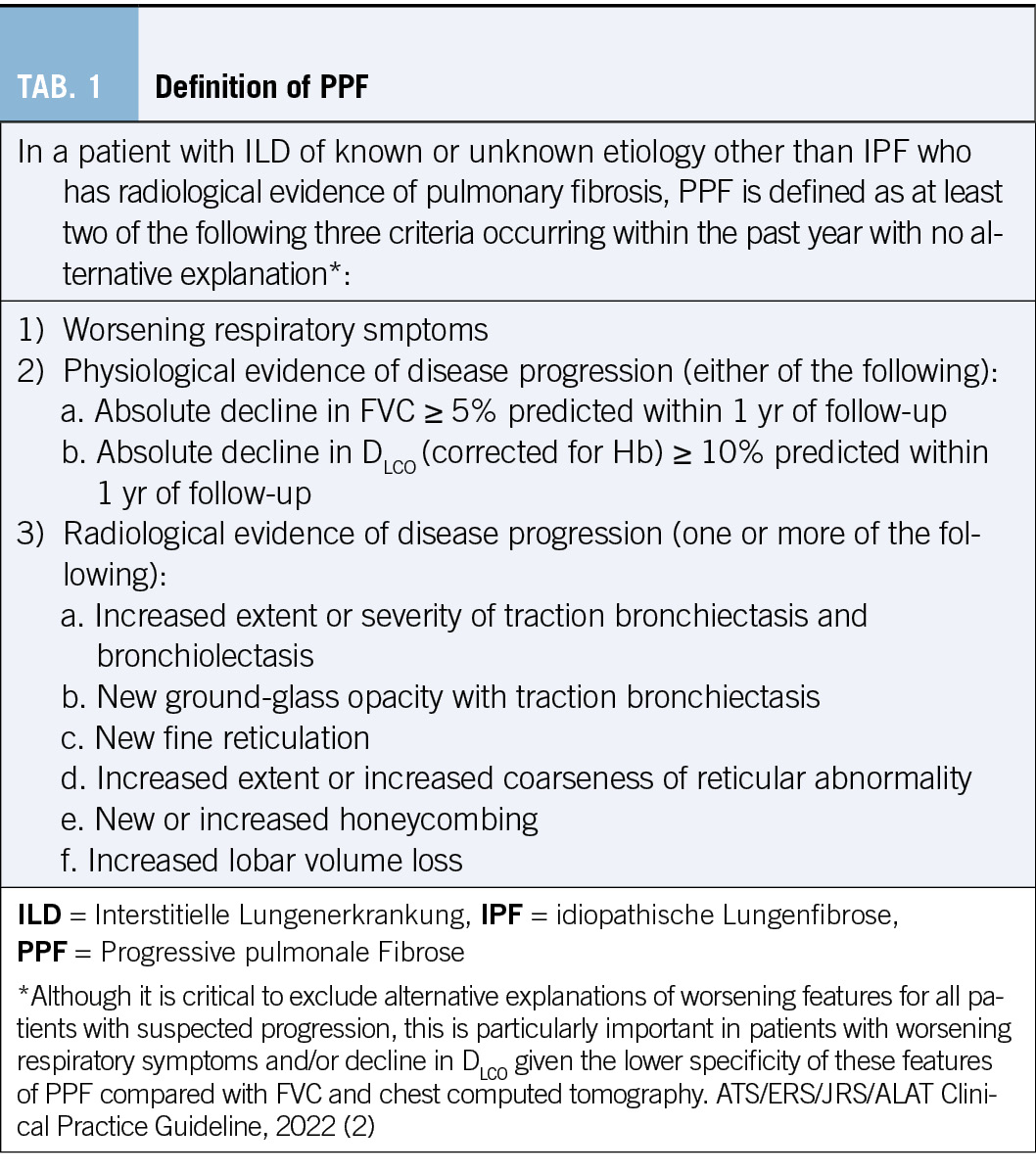
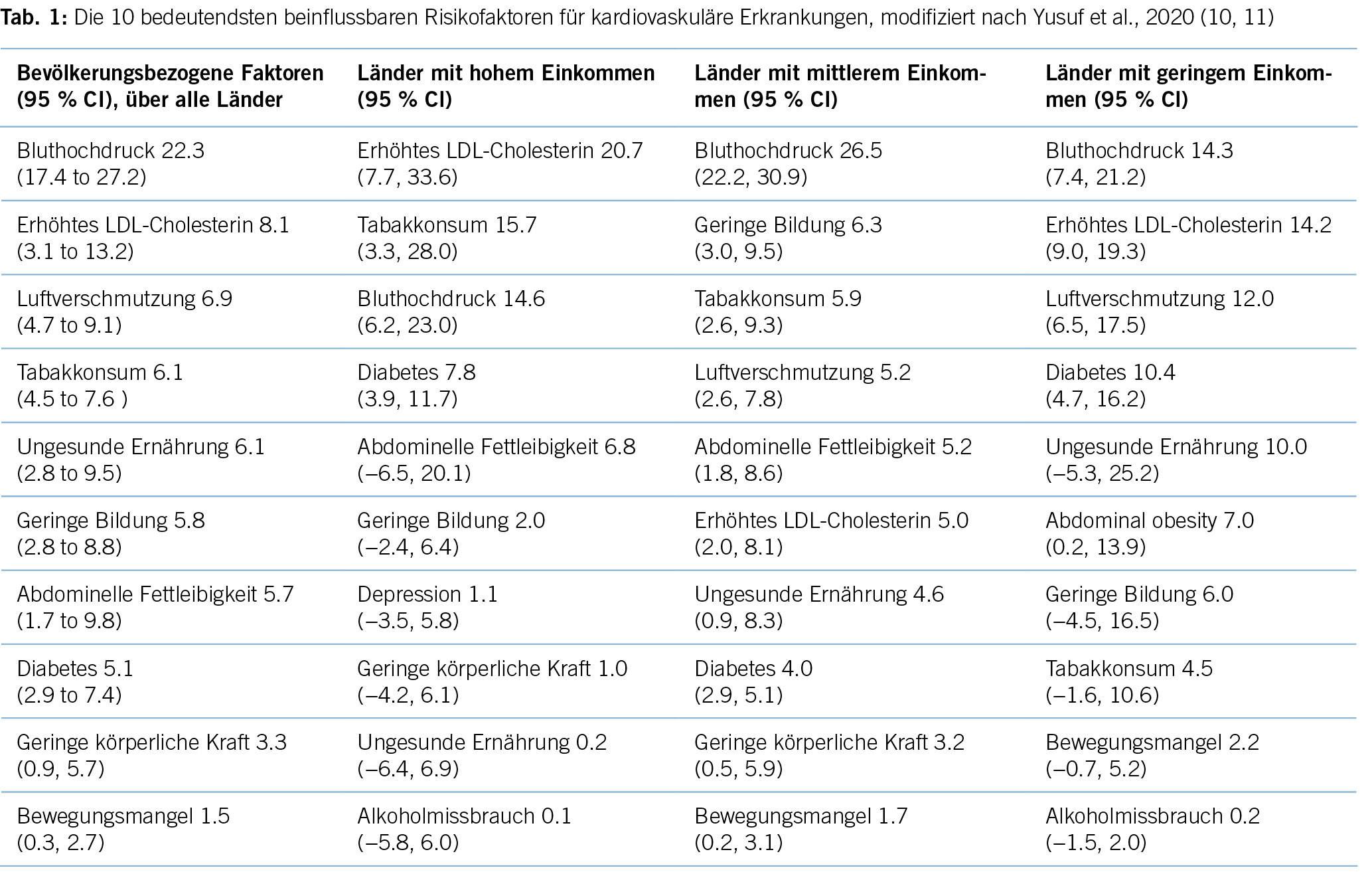
Zahlreiche Studien zeigen eine Diskrepanz zwischen der medizinischen Evidenz respektive den Leitlinienvorgaben und der Primär- und Sekundärprävention in der täglichen Praxis, die dazu führt, dass viele Patient/-innen eine suboptimale Behandlung erhalten (8). Es resultieren unzureichend kontrollierte kardiovaskuläre Risikofaktoren und damit einhergehend vermeidbare kardiovaskuläre Ereignisse respektive eine vermeidbare Krankheits- und Mortalitätslast und direkte und indirekte Kosten (9). Kardiovaskuläre Erkrankungen zeichnen sich dadurch aus, dass die bedeutendsten Risikofaktoren durch Lebensstilmassnahmen oder medikamentöse Interventionen wirksam beeinflussbar sind. Tabelle 1 zeigt die bedeutendsten kardiovaskulären Risikofaktoren, wie sie von Yusuf und Mitarbeitern im Lancet 2020 herausgearbeitet wurden (10).
2.1 Hypertonie
In Ländern mit hohem Einkommen (High-Income) sind 14,6 % aller kardiovaskulären Erkrankungen auf den Risikofaktor Hypertonie zurückzuführen. Für das Risiko, einen kardiovaskulär bedingten Tod zu erleiden, ist der Bluthochdruck sogar der wichtigste Risikofaktor mit etwa 18 % attribuiertem Anteil an den kardiovaskulären Todesfällen. Auch bei Schlaganfällen ist der Blutdruck der führende Risikofaktor mit etwa 34 % aller Schlaganfälle. Für den Myokardinfarkt ist er der zweitwichtigste Risikofaktor mit etwa 11 % zugerechnetem Anteil (10).
Gemäss dem Bundesamt für Statistik (BFS) respektive der Schweizerischen Gesundheitsbefragung (SGB) betrug die Prävalenz der Hypertonie in der Schweizer Bevölkerung im Jahre 2017 17,6 %. Auch hier ist ein Anstieg über die Jahre zu verzeichnen, 1992 waren es noch 14,0 % (12). Zu berücksichtigen ist hierbei, dass die Angaben der Gesundheitsbefragung auf einer Selbstdeklaration («self reported») beruhen und nicht auf tatsächlichen Befunden, etwa aus Hausarztpraxen. Dies dürfte die tatsächliche Prävalenz eher unterschätzen.
Gemäss einer Studie aus dem Jahre 2016 (9), die insgesamt 22 434 Hypertoniker/-innen in Schweizer Hausarztpraxen erfasste, haben 72,7 % dieser Patient/-innen mindestens eine Hypertonie Grad 1, also Werte über 140–159 mm HG. 16,3 % aller Hypertoniker/-innen haben zudem einen Blutdruck in der höchsten Blutdruckkategorie mit Werten von über 180 mm HG systolisch (13).
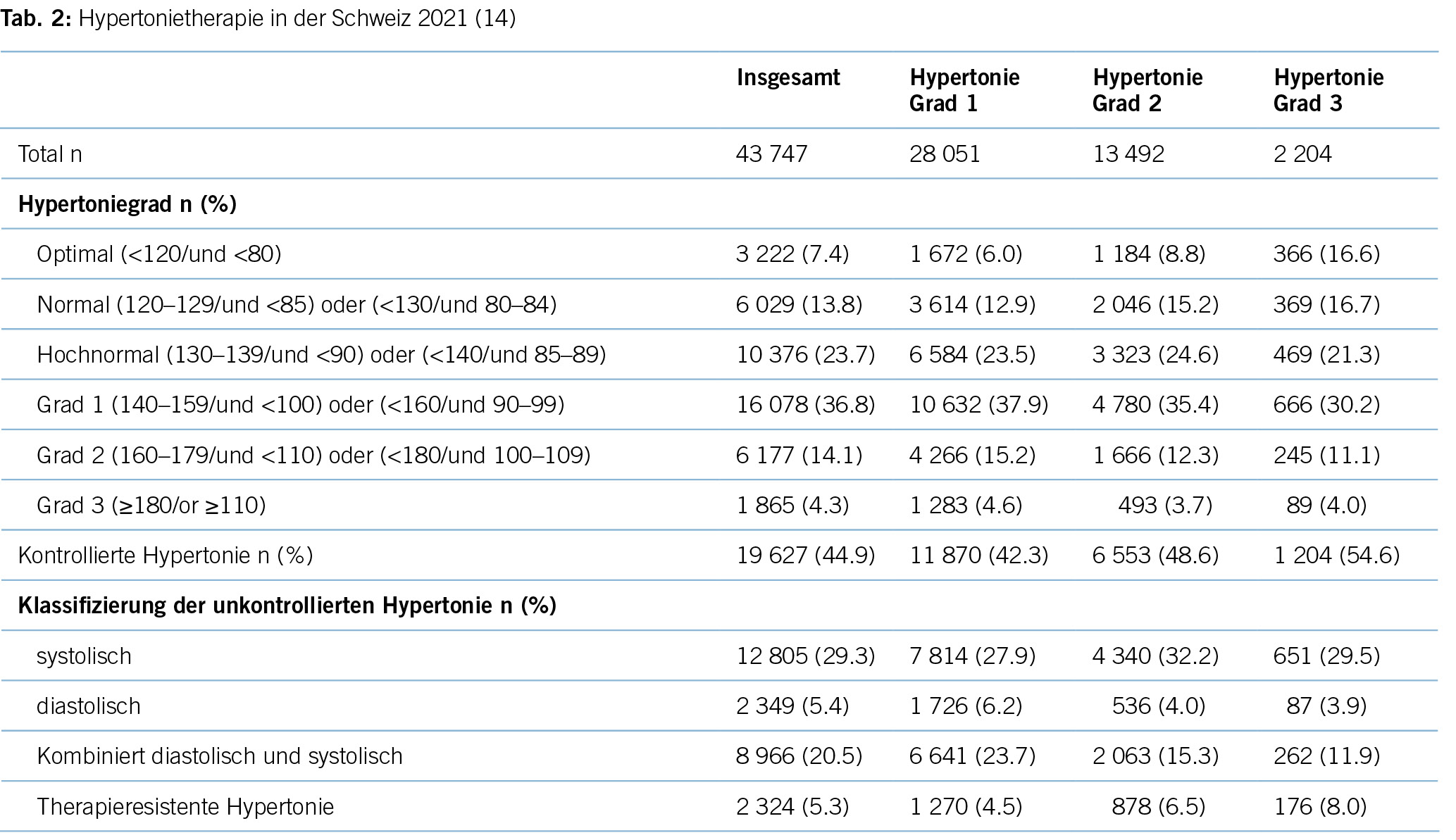
Eine aktuelle Studie aus 2022 zeigt ein ähnliches Bild: 44,9 % der Hypertoniker/-innen sind gemäss der ESC-Leitlinie als «kontrolliert» zu bewerten und im Umkehrschluss mehr als die Hälfte (55,1 %) aller Hypertoniker/-innen nicht im Blutdruckzielbereich (14). Trotz einer Fülle an antihypertensiven Medikamenten ist der Anteil an Patient/-innen, die adäquat für Hypertonie therapiert werden, in der Minderheit. Für die Schweiz bedeutet dies, dass bei einer Bevölkerungszahl von 8,7 Millionen über 843 000 Patient/-innen nicht im Blutdruckzielbereich liegen. Die Gründe hierfür liegen im Unklaren, mangelnde Adherence seitens der Patient/-innen und Multimorbidität mögen einen Teil erklären, wie aber auch im Falle der anderen untertherapierten Risikofaktoren können sie das Ausmass der Untertherapie nicht gänzlich erklären.
Gemäss der Global Burden of Disease Studie und der zugehörigen Datenbank des Institute for Health Metrics and Evaluation (IHME) der University of Washington gingen in der Schweiz im Jahre 2019 allein 310 197 DALYs durch kardiovaskuläre Erkrankungen verloren (15). Das WifOR Institute hat diese Zahlen für 2021 auf 311 332 DALYS hochgerechnet (16). In einem idealisierten Szenario mit Patient/-innen, die sich sämtlich im Blutdruckzielbereich befinden, wären somit etwa 14,6 % oder 45 454 (311 332 x 0,146) verlorene DALYS vermeidbar (15, 16). Dies korreliert gemäss WifOR mit einem vermeidbaren sozioökonomischen Schaden von umgerechnet ca. CHF 7 Mrd. oder etwa 1 % des Bruttoinlandprodukts (BIP) im Jahre 2021.
2.2 Hyperlipidämie
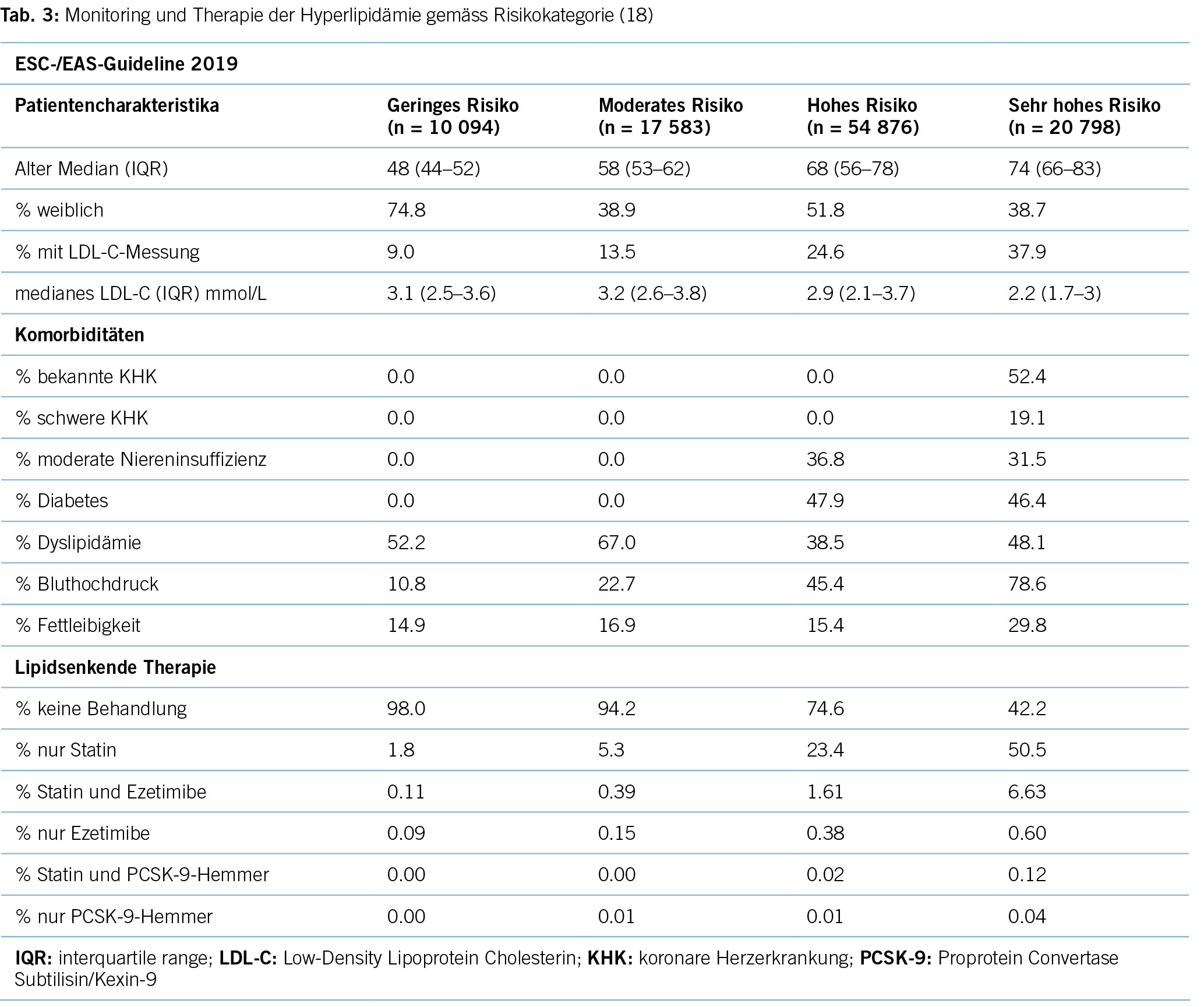
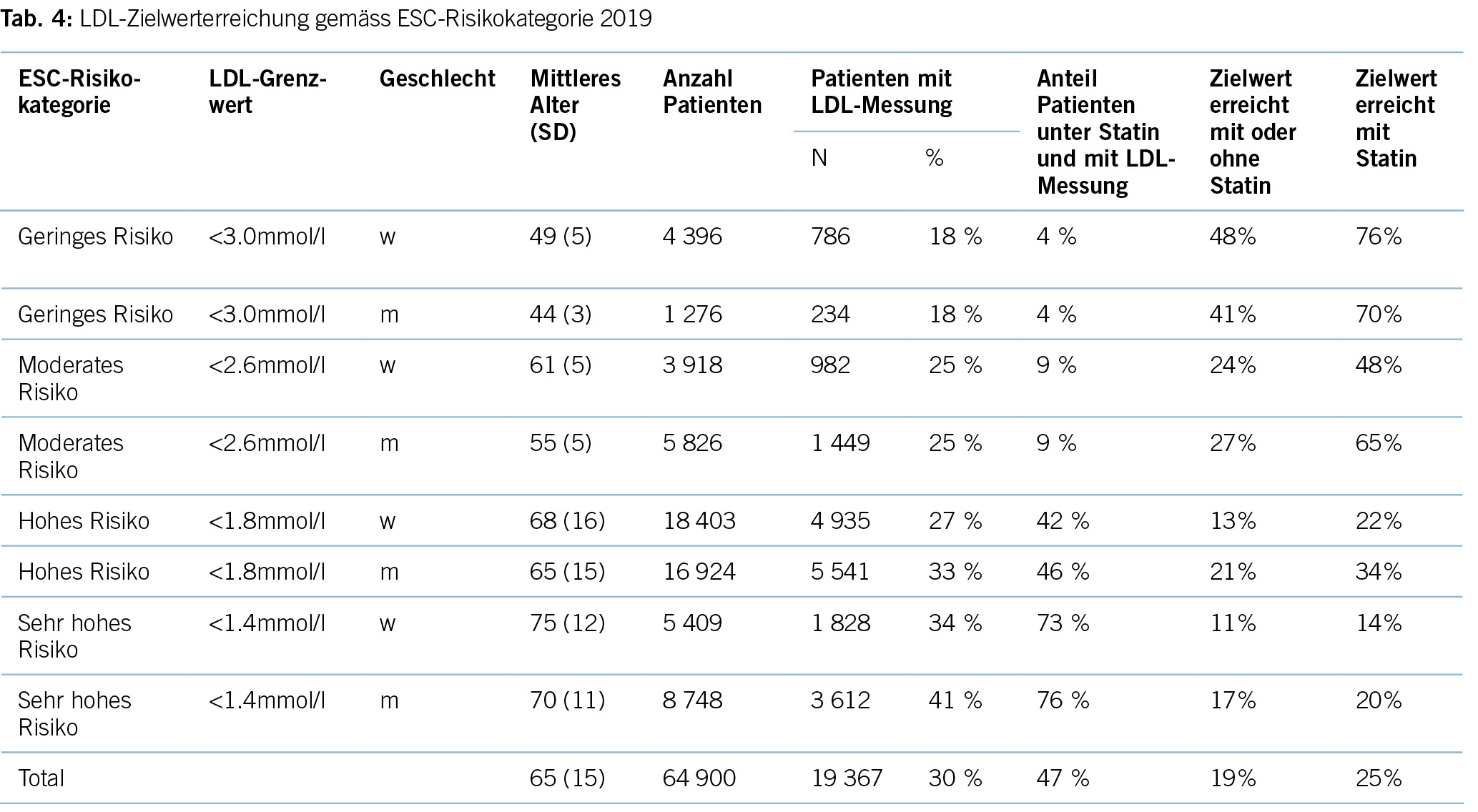
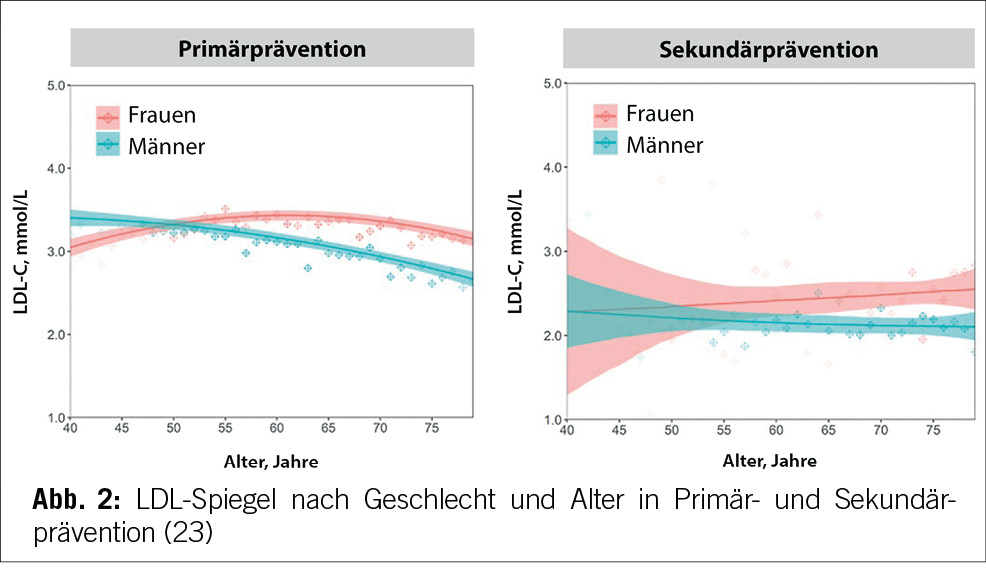
Erhöhtes LDL-Cholesterin ist für 20,7 % aller kardiovaskulären Erkrankungen und rund 5 % aller kardiovaskulären Todesfälle verantwortlich. Im Hinblick auf das Risiko, einen Myokardinfarkt zu erleiden, ist das LDL-Cholesterin der bedeutsamste Risikofaktor mit etwa 12 % attribuiertem Risiko (10). Gemäss BFS und SGB betrug die Prävalenz der Hyperlipidämie (erhöhte LDL-Cholesterinspiegel) in der Schweizer Bevölkerung im Jahre 2017 12,5 %. Bemerkenswert ist insbesondere der Anstieg seit dem Jahre 2002, als in der Gesundheitsbefragung nur 8,8 % von einem erhöhten Cholesterinspiegel berichtet hatten. Innert 15 Jahren war somit ein Anstieg von relativ betrachtet 42 % zu verzeichnen. Dies dürfte neben einem tatsächlich höheren Anteil an Menschen mit erhöhten Cholesterinspiegeln aufgrund einem höheren Anteil älterer Menschen auch einer erhöhten Awareness für die Hyperlipidämie geschuldet sein. Die Europäische Gesellschaft für Kardiologie (ESC) differenziert aufgrund vorliegender Risikofaktoren zwischen verschiedenen kardiovaskulären Risikokategorien, und damit gehen verschiedene Zielwerte im LDL-Cholesterinspiegel einher. Hinsichtlich der Therapie der Hyperlipidämie entsprechend der Risikokategorieeinteilung der ESC-Guideline 2019 zeigt sich folgendes Bild (17) (Tabelle 3): Es wird deutlich, dass bereits das Monitoring der Hyperlipidämie verbesserungsfähig ist: So wird selbst in der höchsten Risikokategorie nur in 37,9 % der Fälle mindestens einmal pro Jahr das LDL-C bestimmt. Immerhin 42,2 % dieser Risikokategorie erhält keine lipidsenkende Therapie. Der mediane LDL-C-Wert in der höchsten Risikokategorie beträgt 2,2 mmol/l (IQR: 1,7–3,0) und liegt damit über dem ESC-Zielwert von 1,4 mmol/l. Betrachtet man die Zielwerterreichung der LDL-Cholesterinwerte gemäss den Zielen der ESC-Guideline 2019, so ergibt sich ein Bild gemäss Tabelle 4. Insgesamt erreichen nur 19 % aller Patient/-innen die Zielwerte, und selbst unter Statintherapie sind dies nur 25 %, was darauf hinweisen könnte, dass im Falle einer lipidsenkenden Therapie zu wenig potente Statine oder eine zu geringe Dosis verabreicht werden. Diese geringe Zielwerterreichung ist nicht nur spezifisch für die Schweiz, ähnliche Werte ergaben auch Studien aus Deutschland, obwohl dort seit mehr als einem Jahrzehnt entsprechende nationale Disease-Management-Programme (DMPs) etabliert sind (19). Zu berücksichtigen ist, dass die ESC-Guideline aus dem Jahre 2019 deutlich tiefere LDL-Grenzwerte vorsieht als die vorangegangene Leitlinie aus dem Jahre 2016. Erfahrungsgemäss gibt es eine Übergangszeit, bis neue Grenzwerte in die tägliche Praxis überführt werden (20). Allerdings zeigten auch die Zielerreichungsgrade mit Berücksichtigung der Zielwerte aus der ESC-Guideline 2016 keine besseren Ergebnisse (18). Eine aktuelle Auswertung aus dem Schweizer FIRE-Netzwerk, einem Forschungsnetzwerk, in das über 700 Hausärzt/-innen ihre Daten aus der elektronischen Krankenakte einspeisen und das Daten bis Ende 2021 einschloss, zeigte überdies keine signifikant besseren Ergebnisse. Die Annahme, dass die Implementierung neuer Zielwerte nur eine gewisse Zeit erfordert, zeigt sich somit nicht bestätigt (21). Bezüglich der Implementierung von Evidenz mittels finanzieller Anreize im Sinne einer leistungsabhängigen Vergütung zeigten Studien, die den Diabetes mellitus adressierten, dass der Höhepunkt der Verbesserung bei einer Incentivierung von Zielerreichungsgraden etwa nach 18 Monaten erreicht wird (22). Bemerkenswert und alarmierend zugleich ist auch die Tatsache, dass bei Frauen das LDL-C als Risikofaktor im Vergleich zu Männern signifikant unterbehandelt wird. Während es bei Blutdruckeinstellung und HbA1c-Werten keine Gendergap gibt, erreichen Frauen sowohl in der Primär- als auch in der Sekundärprophylaxe mit zunehmendem Alter und damit dem Rückgang der protektiven Östrogene klinisch relevant höhere LDL-Spiegel, Abbildung 2 (23). Diese werden aber in der Primär- wie Sekundärprävention nicht adäquat adressiert. Erhöhtes LDL-Cholesterin ist in den High-Income-Ländern für etwa 20,7 % aller kardiovaskulären Erkrankungen verantwortlich (11), was in der Schweiz einem Verlust von 64,445 (0,207 x 311,332) DALYs entspricht. LDL-Cholesterin ist für etwa 16 % aller Herzinfarkte verantwortlich, was etwa 1,000 vermeidbare Herzinfarkte in der Schweiz im Falle einer optimalen Lipidtherapie bedeuten würde. Sozioökonomisch entspricht dies gemäss WifOR 2021 einem vermeidbareren Schaden von 6,4 Mrd. USD (16).
2.3 Diabetes mellitus
Diabetes mellitus ist für 7,8 % aller kardiovaskulären Erkrankungen und 5,9 % aller kardiovaskulär bedingten Todesfälle verantwortlich (10).
4,4 % oder 382 800 Schweizer/-innen über 15 Jahren gaben im Jahre 2017 gemäss Obsan an, an einem Diabetes mellitus zu leiden respektive entsprechende Medikamente einzu- nehmen. Gegenüber 2007 ist dies ein Anstieg um einen Prozentpunkt, was einem relativen Anstieg von etwa 30 % entspricht (24).
Die Therapie des Diabetes mellitus in der Schweiz war immer wieder Gegenstand von Diskussionen. So zeigte beispielsweise eine Studie mit Daten des Krankenversicherers Helsana erhebliches Verbesserungspotenzial beim Monitoring, insbesondere den HbA1c-Kontrollen oder auch den Kontrollen kardiovaskulärer Risikofaktoren wie dem LDL-Cholesterin. Das LDL-Cholesterin wurde beispielsweise nur bei 19,8 % der Patient/-innen einmal jährlich kontrolliert, während dies beim HbA1c in 87,6 % der Fall war (25).
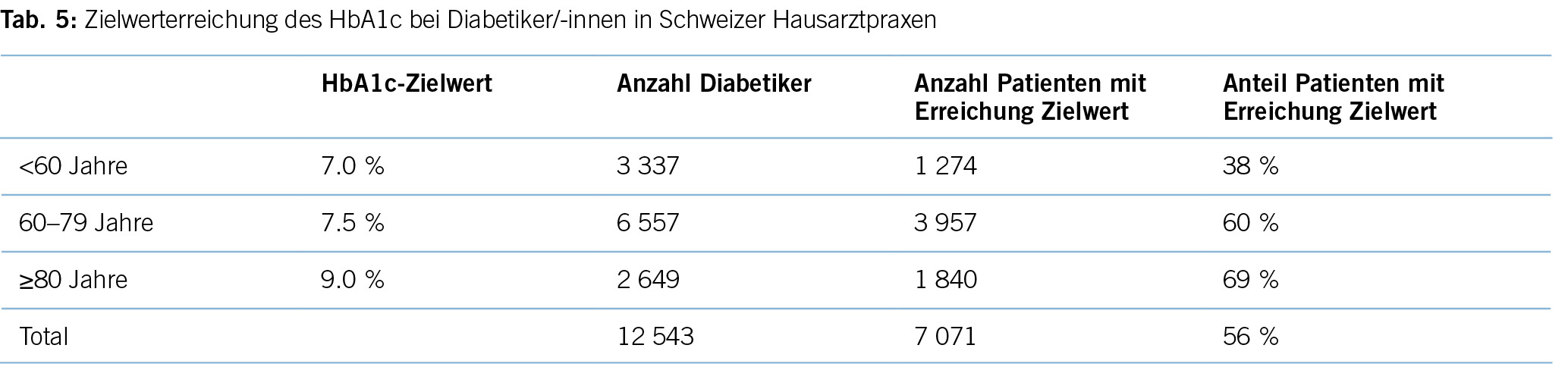
Eine aktuelle Auswertung aus der FIRE-Datenbank mit Routinedaten aus 770 Hausarztpraxen (21) (Tabelle 5) mit 12 543 Patient/-innen zeigt, dass zwar 56 % aller Diabetiker/-innen die altersadaptierten Grenzwerte unterschreiten, dies liegt aber vor allem daran, dass in höherem Lebensalter höhere Grenzwerte angesetzt werden. In der Risikogruppe der unter 60-jährigen, für die ein Grenzwert von 7,0 % im HbA1c gilt, sind es vergleichsweise geringe
38 %. Dies ist umso bedeutsamer, da gerade für diese jüngeren Patient/-innen eine optimale Blutzuckerein- stellung wichtig ist, da sie noch einen grossen Teil der Lebenserwartung vor sich haben.
Den 56 % der Patient/-innen aus Schweizer Hausarztpraxen, die den Zielwert von 7,5 % im HbA1c unterschreiten, stehen 44 % respektive ca. 168 400 (382,800 x 0,44) unkontrollierte Diabetiker/-innen gegenüber, die nicht den altersadaptierten Zielwert erreichen. Etwa 7,8 % aller kardiovaskulären Erkrankungen in High-Income-Ländern sind auf den Diabetes mellitus als Risikofaktor zurückzuführen. Somit sind diesem Risikofaktor der Verlust von 24 283 DALYs (0,078 x 311,332) zuzuschreiben. Diese Zahl reflektiert somit die theoretisch zu gewinnenden DALYs im Falle einer optimalen Therapie aller Schweizer Diabetiker/-innen (24).
2.4 Rauchen
Rauchen ist in den High-Income-Ländern der bedeutsamste modifizierbare Risikofaktor für den Tod insgesamt und für 17,9 % aller Todesfälle und 15,7 % aller kardiovaskulären Erkrankungen verantwortlich (11).
Im Jahr 2017 rauchten gemäss Gesundheitsbefragung 27,1 % der Schweizer/-innen (26). Ein Grossteil der Rauchenden (19,1 % der gesamten Bevölkerung) konsumiert täglich Tabak, ein kleinerer Teil (8,0 %) gelegentlich. Jüngere, nicht repräsentative Daten aus dem Kanton Zürich zeigen eine Zunahme des Tabakkonsums, teilweise auch in anderer Form, etwa Snus (26, 27).
Gemäss einer aktuellen Schweizer Studie von Farcher und Mitarbeitern aus dem Jahre 2023 ist Rauchen für den Verlust von 47 639 DALYs im Jahre 2017 verantwortlich (27).
2.5 Adipositas
Adipositas, definiert als ein BMI von mindestens 30 oder mehr, ist für 6,8 % aller kardiovaskulären Erkrankungen und 11,4 % aller Todesfälle insgesamt verantwortlich, wobei kardiovaskuläre Todesfälle nur zu etwa 5 % der Adipositas zugerechnet werden (11).
In der Schweiz hat der Anteil adipöser und übergewichtiger Personen seit 1992 signifikant zugenommen. Der Anteil stieg von 30,4 % (1992) auf 41,9 % im Jahre 2017. Dabei sind Frauen wie Männer gleichermassen vom Anstieg betroffen, wobei bedeutend mehr Männer (51,0 %) als Frauen (33,0 %) übergewichtig oder adipös sind.
Bedeutsam für das kardiovaskuläre Risiko ist vor allem das abdominelle Fett und somit der Bauchumfang. In der Schweiz wurde im Jahr 2014 bei zwei Drittel aller Personen (66,9 %) ein normaler Bauchumfang gemessen. Bei den Frauen war dieser Anteil etwas höher (68,6 %) im Vergleich zu den Männern (65,0 %). Mit dem Alter (65 bis 75 Jahren) nahm der Anteil Personen mit einem grösseren Bauchumfang auf 31,2 % bei den Frauen und auf 33,4 % bei den Männern zu.
Abdominale Adipositas ist für 6,8 % der kardiovaskulären Erkrankungen in den Industriestaaten verantwortlich, was in der Schweiz für 21 170 (0,68 x 311,332) verlorene DALYs sorgt (11).
2.6 Körperliche Aktivität
Körperliche Aktivität, in einem moderaten Umfang, reduziert das kardiovaskuläre Risiko signifikant (28). Das Bundesamt für Sport (BASPO) empfiehlt allen mindestens 2,5 Stunden Bewegung pro Woche in Form von Alltagsaktivitäten oder Sport mit mindestens mittlerer Intensität bzw. 1,25 Stunden Sport oder Bewegung mit hoher Intensität.
In der Schweiz hat der Anteil der Bevölkerung, der sich gemäss diesen Empfehlungen genügend bewegt, von 62,2 % (2002) auf 75,7 % (2017) zugenommen. Bei den Männern hat der Anteil in diesem Zeitraum um 10,6 % auf 77,8 % zugenommen, bei den Frauen um 15,9 % auf 73,6 %. Der Anteil der Männer, der gemäss den BASPO-Empfehlungen ausreichend körperlich aktiv ist, ist signifikant höher als derjenige der Frauen, allerdings hat sich die Differenz zwischen den Geschlechtern über den Zeitraum von 2002 bis 2017 reduziert. Körperliche Aktivität, wie auch alle anderen kardiovaskulären Risikofaktoren, zeigen eine positive Korrelation mit dem Bildungsniveau. Daten aus Italien zeigen, dass auch nach Korrektur von sozio- demografischen Faktoren gering gebildete Männer ein 21 % und Frauen ein 17 % höheres kardiovaskuläres Risiko im Vergleich zu dem gebildetsten Bevölkerungsanteil haben (29). Global sind etwa 1,5 % aller kardiovaskulären Erkrankungen auf zu geringe körperliche Aktivität zurückzuführen, was auf die Schweiz umgerechnet etwa einem Verlust von 4 669 DALYs entspricht (30). Allerdings dürfte diese pauschale Umrechnung auf die Schweiz den positiven Effekt der körperlichen Aktivität eher unterschätzen, denn diese beeinflusst ausser dem Rauchen praktisch alle modifizierbaren kardiovaskulären Risikofaktoren positiv. Die Steigerung der körperlichen Aktivität stellt daher – neben einer gesunden Ernährung – ein zentrales Ziel in der kardiovaskulären Prävention dar.
3. Das (ökonomische) Potenzial einer optimalen kardiovaskulären Behandlung in der Schweiz
3.1 Direkte und indirekte Kosten der kardiovaskulären Erkrankungen
Offizielle Kostendaten des Bundes zu nicht übertragbaren Erkrankungen (non-communicable diseases, NCD) stammen aus dem Jahre 2011 und stützen sich auf die Arbeit von Wieser und Mitarbeitern (31). Im Jahr 2011 wurden sie demnach auf 52 Milliarden CHF geschätzt was einem Anteil von 80 % der gesamten direkten Gesundheitsausgaben entspricht. Gut 10 Milliarden CHF (15,6 %) davon entfallen gemäss Wieser et al. auf kardiovaskuläre Erkrankungen (32). Hinzu addieren sich indirekte Kosten, die laut Wieser et al. auf 108 % der direkten Kosten geschätzt werden.
Gemäss BFS betrugen die direkten Kosten der Gesundheitsversorgung im Jahre 2021 bereits über 86 Milliarden CHF. Überträgt man die Relationen aus der Arbeit von Wieser et al. auf das Jahr 2021, so ergeben sich mit 15,6 % direkter Kosten der kardiovaskulären Erkrankungen etwa 13,4 Milliarden CHF Kosten im Jahre 2021. Hinzu addieren sich analog 14,4 Milliarden CHF indirekte (108 % der direkten) Kosten, sodass insgesamt von einer Kostenbelastung von 27,8 Milliarden CHF allein durch direkte und indirekte Gesundheitskosten der kardiovaskulären Erkrankungen ausgegangen werden kann. Der Ansatz des WifOR Institute geht über die rein indirekten Kosten, etwa durch Produktivitätsverluste durch den Ausfall einer Arbeitskraft, hinaus und berücksichtigt auch unbezahlte Arbeit, die verringerte Anteilnahme an der sozialen Gemeinschaft etc. Die gesamte sozioökonomische Belastung (socio-economic burden, SEB), die daraus resultiert, beträgt im Jahre 2021 für die Schweiz ca. 26 Milliarden USD (16).
Für die Schweiz geht das WifOR Institute davon aus, dass ein DALY (Abbildung 1) mit einem SEB von CHF 99,417 assoziiert ist und der gesamte sozioökonomische Schaden in der Schweiz durch alle kardiovaskulären Erkrankungen bei umgerechnet circa CHF 31,Mrd. oder 4 % des BIP liegt (16).
3.2 Potenzial für individuelle Gesundheit und vermeidbare ökonomische Belastung
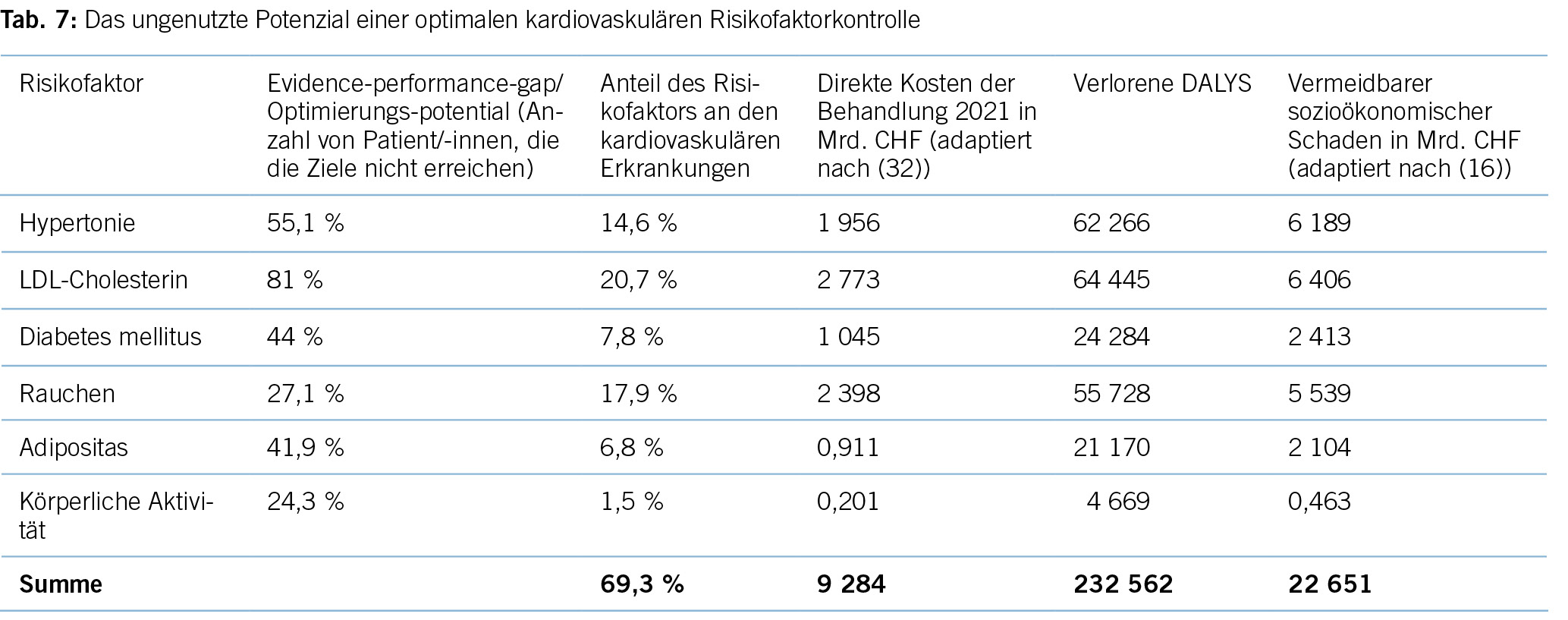
Tabelle 7 zeigt das Potenzial für individuelle Gesundheit, ausgedrückt in DALYs, und der vermeidbare ökonomische Schaden bei einer optimalen Umsetzung der derzeitigen Leitlinienempfehlungen im Hinblick auf die Kontrolle kardiovaskulärer Risikofaktoren in der Schweiz. Eine Änderung des Lebensstils könnte etwa 30 % aller verlorenen DALYs vermeiden, während medikamentöse Interventionen für etwa 70 % des DALY-Potenzials stehen. Auch wenn man keine vollständige Adhärenz auf Patient/-innenseite erwarten kann, ist das Optimierungspotenzial erheblich.
Lösungsansätze
Die Evidenz zur Implementierung von Richtlinien oder Verhaltensänderungen in die tägliche Praxis zeigt, dass die reine Zurverfügungstellung von evidenzbasiertem Wissen nur wenig Effekt hat (33, 34). Aber auch Versorgungskonzepte wie das Chronic Care Modell (CCM), das auf Metaanalysen beruht, die untersucht haben, welche Massnahmen und Rahmenbedingungen die Versorgung chronisch kranker Menschen verbessern, sind ausserhalb von klinischen Studien nie flächendeckend implementiert worden (35–38). Dies, obwohl sich auch im Schweizer Kontext die Verbesserung von Prozessparametern und Outcomes zeigte (39, 40).
Im Vereinigten Königreich (UK) schon 2004 das «Quality and Outcomes Framework» (QOF) initiiert mit dem Ziel, die Versorgung chronisch kranker Menschen zu verbessern. Ein wesentliches Element dabei war die Einführung eines sogenannten Pay-for-Performance (P4P)- Programms für die Hausarztmedizin (41). In diesem P4P-Programm fällt ein relevanter Teil der Vergütung für Hausärzt/-innen auf die Zielerreichung von Qualitätsindikatoren, die die Versorgung chronisch kranker Menschen abbilden. Jedoch können sich bei den Indikatoren trotz regelmässiger Anpassungen Ceiling-Effekte einstellen. Verglichen mit den Morbiditätsdaten anderer europäischer Länder fällt der Rückgang der kardiovaskulären Mortalität in der UK nicht stärker aus, was zu Kritik an diesem zunächst überzeugenden Konzept führte (42). Allerdings bildet das P4P in der UK ausschliesslich Prozessindikatoren ab und keine klinischen Outcomes wie Blutdruck oder LDL-Cholesterinspiegel. Zudem entfallen fast die Hälfte aller generierbaren Vergütungspunkte auf nicht medizinische Faktoren wie die Praxisinfrastruktur, Öffnungszeiten, Erreichbarkeit oder Zufriedenheitsbefragungen der Patient/-innen. Ein RCT in Schweizer Hausarztpraxen mit Prozess- und Outcome-Indikatoren zeigte demnach signifikante Veränderungen bei der gezielten Incentivierung (22, 43).
Conclusion
Kardiovaskuläre Erkrankungen sind die führende Morbiditäts- und Mortalitätsursache in der Schweiz. Es existiert eine umfangreiche wissenschaftliche Evidenz, die in Form von Leitlinien niederschwellig zugänglich zur Verfügung steht. Real Life Daten zeigen aber, dass die vorgegebenen Zielwerte, die ihrerseits mit einem klaren Überlebensvorteil und Verringerung der Morbiditätslast assoziiert sind, häufig nicht erreicht werden.
Multimorbidität mag in einigen Fällen die therapeutischen Optionen limitieren, und auch mangelnde Adherence seitens der Patient/-innen ist ein bedeutsamer Faktor. Beides zusammen mag aber nicht erklären, dass teilweise gerade die Hälfte der Patient/-innen die geforderte Therapie erhält.
Zum Wohle unserer Patient/-innen sind wir Ärzt/-innen gefordert, das aktuelle medizinische Wissen auf die Patient/-innen individuell anzuwenden, denn das ist es, was sie von uns erwarten.
BASPO Bundesamt für Sport
BFS Bundesamt für Statistik
CCM Chronic Care Modell
CVD Cardiovascular diseases, kardiovaskuläre Erkrankungen
DALYs Disability– (oder disease-) adjusted life years, verlorene gesunde Lebensjahre
DMP Disease Management Programme
ESC Europäische Gesellschaft für Kardiologie
GDP Gross domestic product, Bruttoinlandsprodukt
IHME Institute for Health Metrics and Evaluation
LYG Life years gained, gewonnene Lebensjahre
NCD Non-communicable diseases, nicht übertragbare Krankheiten
OBSAN Schweizerisches Gesundheitsobservatorium
QOF Quality and Outcomes Framework
P4P Pay for performance
RCT randomized controlled trial, randomisierte, kontrollierte Studie
SEB Socio-economic burden, sozioökonomische Belastung
SGB Schweizerische Gesundheitsbefragung
WHO World Health Organization
YLD Years lived with disability, durch gesundheitliche Einschränkungen verlorene Lebensjahre
YLL Life years lost, durch Tod verlorene Lebensjahre
Institut für Hausarztmedizin
Universitätsspital Zürich
Pestalozzistrasse 24
8091 Zürich
thomas.rosemann@usz.ch