Ionisierende Strahlung kann Krebs beim Menschen verursachen. Ein konsequenter Strahlenschutz ist deshalb zwingend. Dieser muss auch bei Störfällen von Kernkraftwerken gewährleistet sein. Die vorgesehene
Teilrevision der Kernenergieverordnung wirft diesbezüglich Fragen auf.
Ionisierende Strahlung verfügt über genügend Energie, um Atome und Moleküle zu verändern. Sie kann das Erbgut schädigen und dadurch beim Menschen Krebs verursachen. Das Abschätzen eines dosisabhängigen Krebsrisikos ist allerdings herausfordernd. Entsprechende Grundlagen stammen grösstenteils aus Studien mit relativ kleinen Populationen, welche einer Dosis weit über der gewöhnlichen Strahlenbelastung ausgesetzt waren (1). Gerade das Krebsrisiko bei niedrigen Strahlendosen ist deshalb schwierig abzuschätzen. Es gilt deshalb der Grundsatz, dass es keinen Grenzwert gibt, unterhalb welchem kein Effekt zu erwarten ist. Wenn immer möglich, ist die persönliche Strahlenbelastung durch ionisierende Strahlung zu minimieren.
Risikominimierung durch Strahlenschutz
Die Strahlenschutzverordnung (StSV) schreibt vor, dass die effektive Dosis für eine Person aus der Bevölkerung den Grenzwert von 1 mSv (Millisievert) pro Kalenderjahr nicht überschreiten darf. Spezielle Dosiswerte gelten beispielsweise bei der Exposition gegenüber natürlich vorkommender ionisierender Strahlung, für strahlenexponierte Berufe, bei medizinischen Anwendungen oder bei Stör- und Notfällen. Aber auch hier gilt der Grundsatz der Risikominimierung. Bei Störfällen von Anlagen darf beispielsweise, je nach Wahrscheinlichkeit des Störfalls, eine bestimmte Dosis für Personen aus der Bevölkerung nicht überschritten werden. Bei einem Störfall mit einer Häufigkeit zwischen 10-2 und 10-4 pro Jahr ist dies beispielsweise 1 mSv, bei einem Störfall mit einer Häufigkeit zwischen 10-4 und 10-6 pro Jahr darf die resultierende Dosis maximal 100 mSv betragen. Auch Kernkraftwerke (KKW) müssen so ausgelegt sein, dass die mit der Störfallhäufigkeit verbundenen maximalen Dosiswerte nicht überschritten werden. Kann dieser Nachweis nicht erbracht werden, muss ein KKW vorläufig ausser Betrieb genommen werden. Die damit verbundene Störfallanalyse ist in der Kernenergieverordnung (KEV) geregelt.
Eine Frage der Auslegung
Die KEV weist derzeit eine gewisse Unschärfe in Bezug auf die Störfallanalyse und die damit verbundenen maximalen Dosiswerte auf. Mit einer Teilrevision der KEV soll dieser Interpretationsspielraum aufgehoben werden. Eine entsprechende Vernehmlassung zu den vorgesehenen Änderungen fand Anfang Jahr statt. Klar definierte Ausserbetriebnahmekriterien zur Vorbeugung von Störfällen mit dem Austritt von ionisierender Strahlung in die Umwelt sind in diesem Zusammenhang begrüssenswert und zwingend. Mit der vorgesehenen Teilrevision soll aber neu für Störfälle mit einer Häufigkeit von 10-4 pro Jahr (ein Ereignis in 10 000 Jahren, z.B. ein Erdbeben) ein Dosiswert von 100 mSv als Ausserbetriebnahmekriterium gelten. Eine solche Revision hätte zur Folge, dass die zulässige Strahlendosis für Störfälle mit einer Häufigkeit von bis zu 10-4 pro Jahr gegenüber heute um bis zu einem Faktor 100 steigt. Die Bevölkerung würde so einem zusätzlichen Strahlenrisiko ausgesetzt.
Konsequenter Strahlenschutz, auch bei Störfällen
Störfälle sind keine Alltagsereignisse. Die damit verbundenen Dosiswerte, die nicht überschritten werden dürfen, stellen aber gleichzeitig auch keine Trennlinie zwischen gefährlicher und ungefährlicher Strahlenexposition dar. Auch hier gilt der Grundsatz, dass es keinen Dosiswert gibt, unterhalb dessen ionisierende Strahlung mit Sicherheit keine gesundheitlichen Risiken birgt. Mit diesem Hintergrund stellt sich die Frage, ob ein Dosiswert von 100 mSv für Störfälle mit einer Häufigkeit von 10-4 pro Jahr tatsächlich geeignet bzw. verhältnismässig ist. Zumindest bräuchte es eine klare wissenschaftliche und medizinische Begründung, weshalb der entsprechende Dosiswert neu so hoch angesetzt werden soll. Dabei müsste auch bedacht werden, dass die Bevölkerung nicht homogen ist. Gerade Kinder sind gegenüber einer Strahlenbelastung empfindlicher als Erwachsene (1).
In einer solch heiklen Thematik sind Transparenz und Glaubwürdigkeit die höchsten Güter. Eine entsprechende Rückstellung der vorgesehenen Teilrevision und eine unabhängige Klärung des Sachverhalts wären an dieser Stelle deshalb angebracht.
Dr. Florian Suter
Fachspezialist Prävention und Umwelt
Krebsliga Schweiz
1. Mazzei-Abba, A., Folly, C. L., & Spycher, B. D. Krebsrisiko bei Kindern durch Exposition gegenüber ionisierender S,trahlung. Paediatrica 2018, 29.
Quality of care is the extent to which actual care is in conformity with preset criteria for good care.» Diese noch heute gültige Definition von Qualität in der medizinischen Versorgung von Avedis Donabedian* zeigt auf, dass Qualitätsindikatoren notwendig sind, um die Struktur-, Prozess- und Outcomequalität zu sichern. Strukturen und Prozesse sind jedoch auch massgeblich abhängig von institutionellen und politischen Rahmenbedingungen, die Outcomequalität ergibt sich aus der Kompetenz und Expertise der Fachleute. Dabei ist sowohl die Fähigkeit, die Fertigkeit und das Wissen des Einzelnen zentral aber ebenso die Kompetenz mit anderen zusammenzuarbeiten. Immer mehr rückt die Adhärenz und die Einschätzung des Ergebnisses und der gemachten Erfahrung bei der Therapie der Patientinnen und Patienten (PROM/PREM) in den Fokus. Qualität ist somit ein definitorischer Akt durch Fachgesellschaften selbst, ein Akt der Transparenz und Kommunikation, ein Moment der Planung, Implementierung und Messung aber auch ein Spiegelbild der real existierenden Verhältnisse. Dabei spielen ebenso Qualitätsmanagementprozesse in Spitälern selbst als auch Zertifizierungen eine Rolle, wie auch die öffentlich-rechtlichen Vorgaben von Bund und Kantonen.
Die NSK trägt dem Thema Qualität als ein Schwerpunkt für die Jahre 2017 – 2020 Rechnung. Explizit gehen zwei Projekte dem Thema Qualitätssicherung, Zertifizierung & Outcome nach, die unter dem Lead der SGMO und der KLS stehen. Einerseits geht es um eine Übersicht zu den heute aktuell in der Onkologie bestehenden Qualitätssicherungs- und Qualitätsentwicklungsmethoden (NSK 3.2.1), andererseits um die Qualitätssicherung für regionale Behandlungsnetzwerke (NSK 3.2.2).
Dr. Hermann Amstad ist von der KLS in Absprache mit der SGMO mandatiert worden, die erwähnte Situationsanalyse unter Einbezug einer Expertengruppe zu realisieren. Der Bericht soll zu zwei Themenkreisen Informationen liefern. Einerseits zur Ist-Situation der Zertifizierungen und der Qualitätsentwicklung in der Schweiz. Andererseits zu den in der Schweiz existierenden Vorgaben zur Qualitätssicherung im Bereich «Versorgungspfade in onkologischen Netzwerken», die wissenschaftlich fundiert, von den Stakeholdern akzeptiert und in der Praxis umsetzbar sind.
Der Bericht soll Ende Jahr vorliegen und Ausgangspunkt für weitere Projektschritte und mögliche Aktivitäten sein:
1. Konkretisierung einer Qualitätssicherung für Versorgungspfade in onkologischen Netzwerken.
2. Klärung durch die SGMO, die Arbeitsgemeinschaft Krebszentren und weitere DKG zertifizierte Organ- und Krebszentren zum Vorgehen in Sachen «Swissness» der DKG Zertifizierung, also einer Form von Zertifizierung, die auf dem deutschen Modell der DKG basiert, gleichzeitig jedoch den Besonderheiten der föderalen schweizerischen Strukturen und Sprachkulturen Rechnung trägt (NSK 4.1.1).
3. Diskussion in der SGMO im Kontext Smarter Oncology/Choosing wisely.
Die beschriebenen Aktivitäten sind teilweise eng verknüpft mit anderen Projekten der NSK, beispielsweise zur Frage nach Prinzipien und Anforderungen an Tumorboards (NSK 3.3) sowie zu Guidelines und Behandlungsrichtlinien. Dazu ist insbesondere auch auf die Arbeiten der SAQM/FMH zum sektor- und berufsgruppenübergreifenden Behandlungspfad Kolonkarzinom hinzuweisen (NSK 3.1). Als Moment der externen Qualitätssicherung ist zudem auf die Einführung des Krebsregistrierungsgesetzes (NSK 7.1) und die Arbeiten an den Registerdaten zur Behandlungsqualität und Datenverknüpfung (NSK 7.2) hinzuweisen. Last but not least sei in diesem Zusammenhang auch auf die Stärkung der Fachkompetenz der Gesundheitsfachleute hingewiesen (NSK 5.2.1 & 5.2.2).
* Avedis Donabedian: The Definition of Quality … Band 1, Health Administration Press, 1980, ISBN 0-914904-48-5.
La grippe saisonnière est une maladie infectieuse virale aiguë due aux virus influenzae A/H1N1, A/H3N2 et influenzae B. Encore trop souvent considérée comme une affection bénigne par le plus grand nombre, elle est très contagieuse et associée à des complications parfois graves. La grippe est à l’ origine de 1000 à 5000 hospitalisations et 1500 décès chaque année en Suisse dont 90 % chez les 65 + (www.bag.admin.ch). Cependant, la mortalité ne représente que la face émergée de l’ iceberg. La grippe favorise également la décompensation de maladies chroniques et peut agir comme un facteur de déclin fonctionnel notamment chez les patients les plus âgés et fragiles (1, 2).
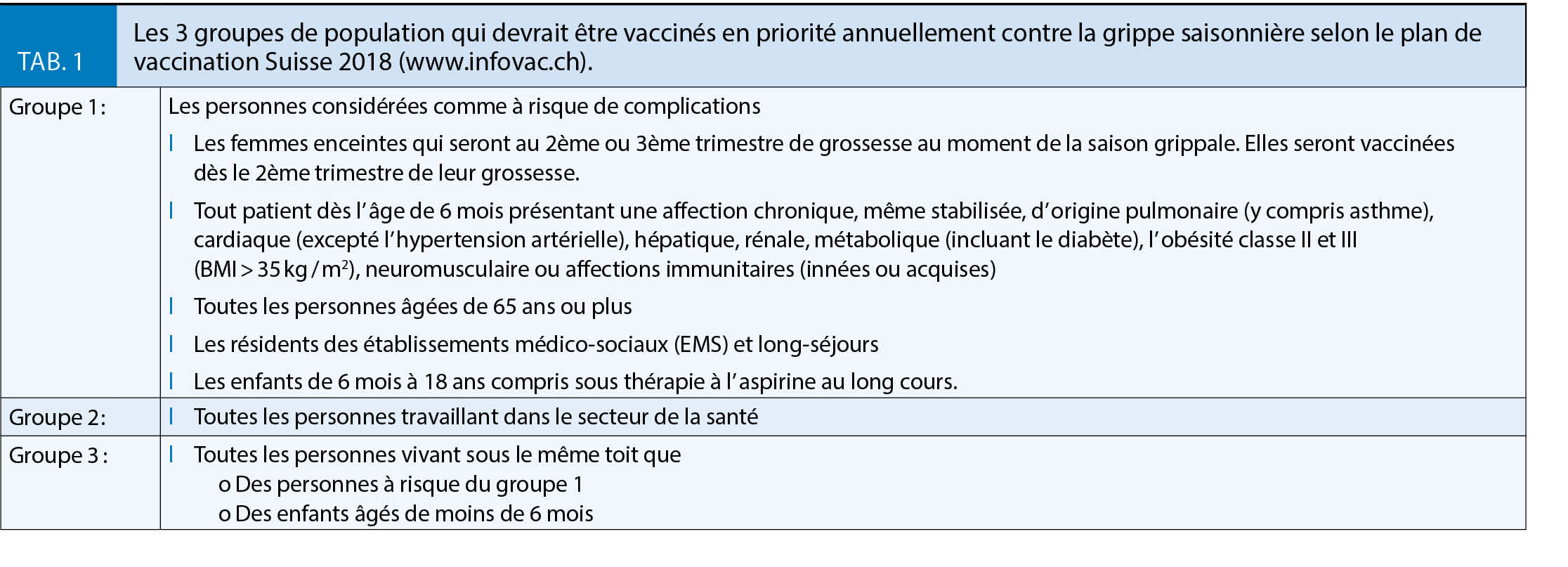
La prévention vaccinale est la mesure la plus efficace pour lutter contre la grippe, même si l’ exacte efficacité des vaccins actuels reste débattue (3-6). Les recommandations en matière de vaccination contre la grippe saisonnière sont inchangées depuis 2013 (www.infovac.ch). La vaccination est recommandée annuellement chez les personnes dites à risque accru de complications, leurs proches, et chez les professionnels de la Santé (tab. 1) (1). Il n’ y a à ce jour pas d’ évidence d’ une réduction de l’ efficacité avec ce schéma de vaccination (7). Certains bénéfices en matière de couverture antigénique ont même été suggérés (8).
La grippe en chiffres
En Suisse, la grippe est à l’ origine de 112 000 à 275 000 consultations médicales chaque année (selon le système de surveillance Sentinella). Durant la saison grippale 2017/18, sur les 15 semaines qu’ ont duré l’ épidémie (sur la période du 1er octobre 2017 au 21 avril 2018), les principaux virus circulants étaient l’ influenzae B du lignage Yamagata (66 %) et le virus A/H1N1 pdm 09 (23 %) ; les virus A/H3N2 et B du lignage Victoria n’ ont été que sporadiquement isolés.
Durant toute l’ épidémie, le taux de consultations hebdomadaires était au-dessus du seuil épidémique de 68 consultations pour 100 000 habitants avec deux pics au cours de la deuxième et de la quatrième semaine de 2018 (358 et 352 consultations/100 000 habitants) qui étaient inférieurs à ceux mesurés en 2008/09, 2012/13, 2014/15 et 2016/17.
Si l’ incidence était maximale chez les enfants de 0 à 4 ans (6258 consultations/100 000 habitants), les 65 + était la classe d’ âge la moins infectée avec tout de même 2549 consultations/100 000 habitants. Avec près de 4 % de la population ayant consulté un médecin de premier recours durant la période épidémique (3950 premières consultations/100 000 habitants), il s’ agit de la valeur la plus élevée enregistrée en Suisse depuis 2000 (+ 46 % de la moyenne des 10 dernières saisons). La principale raison est avant tout la longueur exceptionnelle de l’ épidémie ; en moyenne au cours des dix dernières années, la durée des saisons grippales était de 10,5 semaines (www.bag.admin.ch).
Chez les 65 +, le nombre de décès n’ a que très légèrement dépassé les valeurs attendues au début mars 2018. Chaque année, cette surmortalité témoigne de l’impact de l’ épidémie dans cette population et du risque d’ évolution grave chez les personnes vulnérables. Parmi l’ ensemble des cas de grippe déclarés, 7 % appartenait au groupe des personnes présentant un risque accru de complication et 30 % pour les 65 +. Une pneumonie a été diagnostiquée dans 4 % de l’ ensemble des cas ; le plus souvent parmi les plus âgés (12 %) et le plus rarement chez les enfants ≤ 4 ans (1 %). Près de 1 % des personnes suspectés d’ affection grippale et 9 % de celles avec une pneumonie ont été hospitalisées. La proportion la plus élevée d’ hospitalisations pour suspicion de grippe était enregistrée chez les 65 + (3 %). Les 65 + enregistraient aussi le plus haut taux de détection intra-hospitalière (54 % vs. 33% chez les 30-64 ans) (www.bag.admin.ch).
Durant la saison 2017/18, environ 7 % des personnes déclarées pour suspicion de grippe avec un statut vaccinal connu avaient été préalablement vaccinées. Cette proportion était plus importante dans les groupes chez qui l’ OFSP recommande la vaccination (tab. 1), avec 31 % chez les 65 + et 39 % chez les personnes à risque accru de complications témoignant du peu d’ efficacité du vaccin. Ces données ont été confirmées à l’ échelle internationale. Un traitement antiviral, dans la plupart des cas par un inhibiteur de la neuraminidase, a été administré chez 1 % des personnes déclarées avec une grippe ; 11 % ont reçu un traitement antibiotique, probablement en raison d’ une surinfection bactérienne (www.bag.admin.ch).
La grippe est contagieuse avant les symptômes et parfois même asymptomatique
La grippe se transmet par contact direct avec une personne infectée (éternuement, toux jusqu’ à 1 mètre), notamment dans des espaces clos. Les virus grippaux peuvent aussi rester vivants jusqu’ à 48 heures sur des surfaces inertes. Comme il a été estimé qu’ un individu adulte peut toucher jusqu’ à 40 fois son visage par heure avec ses mains, les manipulations d’objets et les contacts avec des surfaces inertes « contaminés » (tab. 1: poignées de portes, bouton d’ ascenseur, rampe d’ escalier, billet de banque, etc.) sont une réelle voie de transmission à ne surtout pas banaliser (9). Les personnes contaminées peuvent transmettre les virus de la grippe à d’ autres même si elles ne se sentent pas (encore) malades (9) sur leur lieu de travail, à la maison et/ou dans les institutions de santé telles que les EMS ou les hôpitaux.
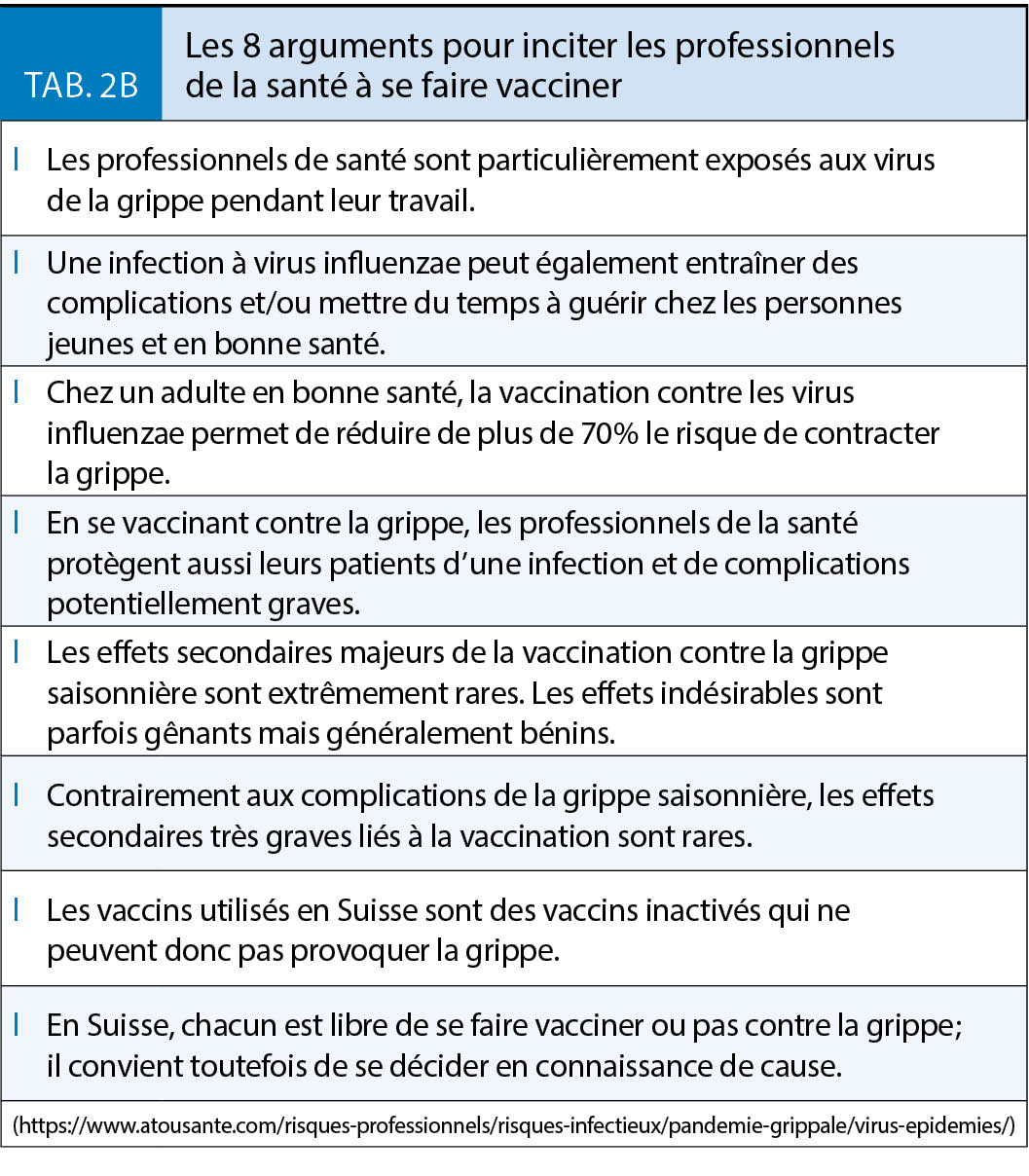
Près d’ un tiers des personnes infectées par un des virus de la grippe saisonnière ne présente aucun des symptômes spécifiques (10). Ces personnes peuvent néanmoins transmettre le virus. La vaccination contribue fortement à diminuer le risque de contagion (11). Les professionnels de la Santé sont parmi les plus fortement exposés au risque de contracter la grippe. De plus, les arrêts de travail pour maladie qui en résultent impliquent souvent une charge de travail supplémentaire pour les collègues en période épidémique et/ou des contraintes de réorganisation en rapport avec le recours à du personnel intérimaire notamment dans les EMS et les hôpitaux (12).
La grippe en clinique
Après contamination, les symptômes grippaux apparaissent généralement en un à trois jours. La grippe saisonnière se manifeste par une sensation de malaise général, une brusque poussée de fièvre, des frissons, des maux de tête, des arthro-myalgies, une perte d’ appétit et des vertiges. La seconde phase se caractérise par l’ intensification des symptômes respiratoires (toux sèche, maux de gorge, enrouement, rhinite). La fièvre dure en générale 3 à 8 jours et la convalescence 7 à 15 jours mais peut se prolonger au-delà (13). Cependant chez les personnes âgées et/ou celles présentant des affections chroniques, la grippe est loin d’ être une maladie bénigne et peut s’ accompagner des complications (14). Les plus fréquentes sont les pneumonies infectieuses. Primaires, elles sont dues à la virulence directe du virus de la grippe ; secondaires, à une surinfection bactérienne (14).
Les pneumonies au cours de la grippe
Deux formes de pneumonies peuvent survenir au cours de la grippe. La pneumonie grippale, d’ origine virale, se manifeste par une détresse respiratoire aiguë quelques jours après le début de l’ infection. Les pneumonies bactériennes se développent généralement plus secondairement (13, 15, 16).
La pneumonie grippale
La pneumopathie à virus influenzae est une complication majeure mais rare. Elle concerne préférentiellement les enfants de moins de 2 ans et les 65 +. Généralement bénigne et de courte durée chez l’ enfant, elle est gravissime chez l’ adulte (13).
Elle se caractérise par une toux fébrile suivie d’ une dyspnée, puis l’ apparition d’ une cyanose. La radiographie du thorax va montrer un infiltrat nodulaire ou réticulo-nodulaire avec ou sans foyer de condensation. Le scanner retrouvera des images de condensations péribronchiques et / ou sous pleural ainsi que des images en verre dépoli. L’ aspect radiologique et les caractéristiques cliniques peuvent mimer en tout point un syndrome de détresse respiratoire aiguë. La pneumonie grippale correspond à une atteinte directe du parenchyme pulmonaire par le virus grippal avec soit atteinte des alvéoles avec œdème hémorragique intra-alvéolaire soit de l’ inter-stitium et induction d’ une fibrose aiguë (avec généralement des séquelles respiratoires). Dans les formes les plus graves, une prise en charge en milieu de réanimation est le plus souvent nécessaire. Parfois une myocardite est associée. Dans sa forme maligne, elle survient en général dans les 24 premières heures de l’ infection mais peut survenir jusqu’ à 10 jours après le début de la grippe. Le risque de développer une pneumopathie grippale résulte d’ une réponse complexe impliquant un système immunitaire sidéré d’ une part et les caractéristiques du virus d’ autre part.
Les sujets âgés, particulièrement les plus dépendants et vivant en institution, ainsi que ceux ayant des comorbidités cardiovasculaires et/ou respiratoires constituent habituellement le groupe le plus à risque. Quoi qu’ il en soit, selon les sous-types de virus, les groupes les plus à risque peuvent varier, comme par exemple au cours des pandémies de 1918 et 2009 où les sujets jeunes étaient préférentiellement atteints. La mortalité reste élevée, de l’ ordre de 30 % avec un décès survenant généralement dans les 4 jours (13), notamment en cas de coinfections par Staphylococcus aureus ou Streptococcus pneumoniae (17).
Les pneumonies bactériennes
Les surinfections bactériennes s’ observent dans toutes les tranches d’ âge et représentent la complication la plus fréquente (16-18). Les lésions provoquées par le virus influenza favorisent la prolifération bactérienne dans le tractus respiratoire (16). Les germes les plus fréquemment isolés sont S. aureus, S. pneumoniae et Hæmophilus influenzae. Contrairement à la pneumonie à S. aureus, les pneumonies à pneumocoque et Haemophilus surviennent généralement plus tard, entre 2 et 3 semaines après le début des symptômes grippaux, et peuvent être traitées en ambulatoire selon les mêmes modalités et recommandations de prise en charge qu’ une pneumopathie aiguë communautaire non compliquée. Les surinfections sont également favorisées par des lésions préexistantes associées à certaines pathologies chroniques notamment. Cela explique pourquoi la grippe est une maladie grave chez les 65 +, les insuffisants respiratoires et/ou cardiaques, les diabétiques et est alors associée à une morbimortalité très supérieure. La ré-analyse des pièces d’ autopsie pulmonaires des personnes décédées de la pandémie grippale de 1918 a confirmé que la grande majorité des décès alors n’ avait pas été directement liée à la seule virulence du virus influenza, mais bien à des surinfections par S. pneumoniae et S. aureus (19). Si l’ évolution est le plus souvent favorable sous une antibiothérapie adaptée, le pronostic dépend cependant avant tout du terrain sous-jacent et est bien plus sombre chez des patients âgés, fragiles, polymédiquées et polymorbides.
Prévention : la vaccination, recommandée chaque année, est le moyen le plus efficace
La vaccination reste la prévention la plus simple, efficace et économique chez les personnes à risque de complications (tab. 1), celles qui s’ en occupent ou leur entourage, dans les milieux de soins, les collectivités, et la vie courante. La période idéale de vaccination va de mi-octobre à début décembre. Les autres mesures préventives, notamment les règles d’ hygiène, même si elles sont indispensables, restent un complément à la vaccination antigrippale mais ne peuvent la remplacer. En l’ absence de vaccin ou de traitement spécifiques des autres infections respiratoires hivernales, les masques, les appareils de protection respiratoire et l’ hygiène des mains ainsi que les mesures barrières (isolement « gouttelettes », éloignement social) en structure institutionnelle mais aussi en ambulatoire sont de ce fait les seules armes efficaces (20-22).
En matière de recommandation vaccinale, chez l’ adulte, il n’ y a pas d’ arguments cliniques particuliers à privilégier un vaccin trivalent (3 souches grippales – pour la saison 2017/18 : A/H1N1pdm09 = A/Michigan/45/20154, A/H3N2 = A/Hong Kong/4801/2014, B Victoria = B/Brisbane/60/2008) à un vaccin tétravalent (4 souches grippales – pour la saison 2017/18 Trivalent + B Yamagata = B/Phuket/3073/2013). Tous les vaccins autorisés en Suisse sont inactivés et exempts de mercure et d’ aluminium. Les vaccins disponibles et autorisés pour les adultes sont : Agrippal®, Fluarix®, Influvac® et Mutagrip®. Fluarix Tetra® est un vaccin quadrivalent. De plus, le vaccin Fluad® qui contient un adjuvant (MF59C) qui en renforce l’ efficacité (23) est plus particulièrement recommandé chez les 65+ (www.sevaccinercontrelagrippe.ch) (24). Si les vaccins sont disponibles pour tous, la priorité est la vaccination des personnes appartenant à un groupe à risque de complications (tab. 1) (www.infovac.ch).
La composition des vaccins est déterminée chaque année en février par l’ Organisation mondiale de la santé (OMS). Depuis 2013-2014 dans l’ hémisphère nord, l’ OMS formule également des recommandations sur la composition de vaccins quadrivalents. Pour la saison 2018/2019 la composition du vaccin trivalent a été modifiée en ce qui concerne les souches A/H3N2 (A/Singapore/INFIMH-16-0019/2016) et B-Victoria (B/Colorado/06/2017) afin de mieux couvrir les virus en circulation. La souche supplémentaire influenzae B contenue dans le vaccin tétravalent est inchangée. Sans adjuvant, les vaccins sont disponibles depuis la fin du mois de septembre.
Globalement, la vaccination permet de réduire de 70 % le risque de grippe chez un adulte en bonne santé lorsque les souches vaccinales correspondent bien aux souches circulantes (ce qui n’ a pas été le cas notamment durant la saison 2015/16) (5). L’ âge et les capacités immunitaires du vacciné (24) contribuent à expliquer pourquoi la protection vaccinale s’ abaisse à 30-40 % chez les seniors (3, 4). En milieu institutionnel, la vaccination du personnel et des résidents conduit à une réduction de 46 % des pneumonies, de 45 % des hospitalisations, et des décès dus à une grippe ou une pneumonie de 42 % (25, 26). Chez 5 % des personnes vaccinées, des réactions similaires aux symptômes grippaux sont décrites. Elles ne sont pas la grippe, mais le témoin de la réponse immunitaire à la vaccination. Si les adjuvants améliorent l’ immunogénicité des vaccins, ils augmentent aussi la réactogénicité, qui se résument le plus souvent à des réactions au point d’ injection plus intenses mais souvent bénignes (1).
Durant la saison grippale 2017/18, la couverture des souches circulantes par le vaccin trivalent était faible (29 %) compte tenu de la prépondérance du virus B lignage Yamagata contenu uniquement dans le vaccin tétravalent (95 % de protection). Si l’ efficacité vaccinale a été estimée à 25-52 % selon la catégorie d’ âge (souche A/H1N1pdm09 : 55-67 % ; virus B : 36-55 %), les vaccins trivalents ont néanmoins démontré une efficacité contre l’ influenzae B/Yamagata en raison d’ une protection croisée entre les lignages (49- 77 %) (www.bag.admin.ch). Ces données proviennent des USA car aucune étude d’ efficacité n’ a été réalisée en Suisse.
De façon intéressante, les effets immunomodulateurs de la VitD ont été considérés dans la prévention de la grippe et des infections respiratoires saisonnières (27). Dans un essai randomisé contrôlé en long séjour, Ginde et al. ont montré qu’ une supplémentation par 100 000 UI/mois de VitD réduisait l’ incidence des infections respiratoires aiguës (2) comparativement à une supplémentation selon les recommendations habituelles de 400-1000 UI/jour (28). Si les effets anti-infectieux de la VitD sont de mieux en mieux documentés, aucune donnée actuellement ne confirme un effet de la supplémentation en VitD sur l’ amélioration de l’ immunogénicité des vaccins antigrippes (29).
Le traitement : la place des antiviraux
Des antiviraux contre la grippe sont disponibles en Suisse et leur utilisation permet d’ éviter des complications sévères et des décès dans les situations à risque. Dans l’ idéal, ils doivent être administrés au plus tôt après le début de la maladie. Le traitement empirique des patients suspects d’ avoir une grippe n’ est habituellement pas recommandé. Un traitement antiviral est indiqué pour les patients dont la maladie respiratoire est sévère, durant la période d’ épidémie avec des symptômes grippaux de moins de 48 heures (30).
Les inhibiteurs de la neuraminidase (oseltamivir, zanamivir) limitent la diffusion des virus en dehors des cellules infectées et les inhibiteurs de la protéine M2 (amantadine, rémantadine) limitent la pénétration du virus dans la cellule. Ils réduisent efficacement les complications et plus généralement l’ évolution des symptômes. Si la grande majorité des virus y sont encore sensibles, certaines mutations conduisent à des résistances (neuraminidase : H275Y et E119V ; gène de la protéine M2 : Ser31). Les taux de résistance pour les virus grippaux en circulation sont sous étroite surveillance. L’ OMS peut fournir en temps réel les informations relatives à l’ utilisation possible dans la prise en charge thérapeutique ou prophylactique (par ex. épidémie en communautés fermées, institution, etc.) (30, 31). Ainsi, durant la saison 2017/2018, un seul des 91 virus analysés dans le réseau Sentinella présentait une résistance contre l’ oseltamivir et globalement les cas de résistance sont rares (Europe < 0.3 % et USA : 1 % des A/H1N1pdm09, et 0 % pour les autres virus) (www.bag.admin.ch).
Conclusion
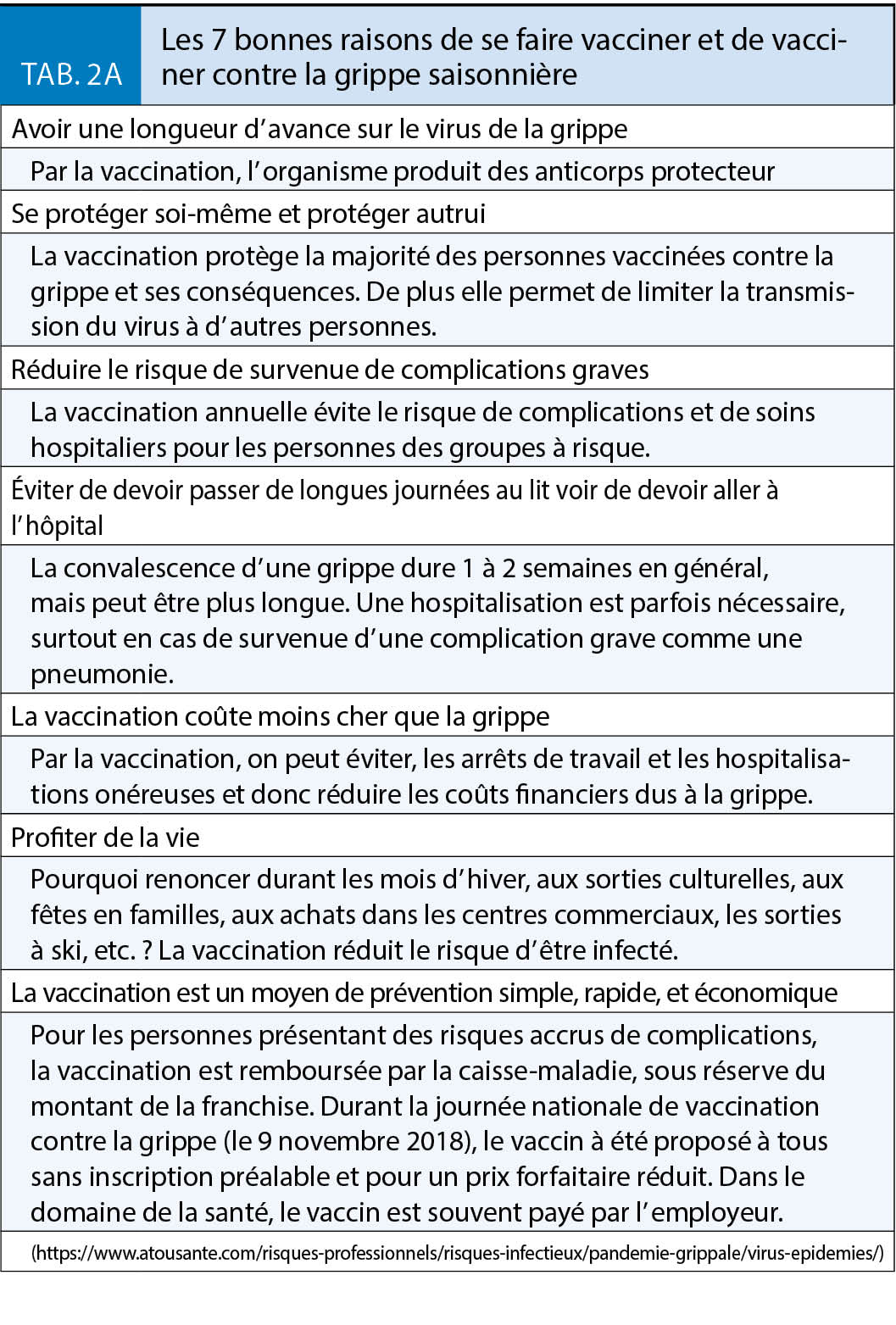
La grippe est l’ infection qui, en Suisse, tue chaque année le plus de personnes et notamment les 65 +. La vaccination chez les personnes âgées est une priorité tout comme chez tous les adultes du groupe 1 et les professionnels de la Santé. Selon une enquête de l’ OFSP (n = 3605) ciblant les personnes qui auraient dû être vaccinées durant la saison 2017/18, seuls 32 % des 65 + l’ étaient et 25 % des porteurs de maladies chroniques. Si 18 % des professionnels de la santé s’ étaient fait vacciner en 2014/15, 21 % en 2015/16 et 25 % en 2016/17, le taux de couverture s’ est abaissé à 20 % durant la saison 2017/18. Pour la première fois, cette enquête a aussi ciblé les personnes en contact régulier avec une personne à risque et le taux de vaccination n’ était que de 7 % (www.bag.admin.ch). Si les mesures de protection individuelles (port de masque et hygiène des mains) sont un bon complément, il faut redoubler d’ effort pour améliorer les taux de couverture vaccinale chez les patients à risque et les professionnels de santé (tab. 2A et 2B).
Dr. med. Pierre-Olivier Lang, PhD
Genolier Klinik und Montchoisi Klinik
Route du Muids 3
1272 Genolier
plang@genolier.net
plang@genolier.net
L’ auteur n’ a aucun conflit d’ intérêt en relation avec cet article.
La grippe est une infection virale aiguë très contagieuse.
La grippe provoque des épidémies annuelles avec un pic hivernal.
La grippe est un problème majeur de santé publique notamment dans les populations dites à risque ce qui inclut toutes les personnes ≥ 65 ans.
Dans un tiers des cas, la grippe reste asymptomatique mais la personne est contagieuse et peut transmettre le virus à tout son entourage.
Les professionnels de la Santé sont particulièrement à risque d’ être infectés et de transmettre la grippe.
La vaccination est actuellement encore le moyen de prévention le plus efficace ; les mesures de protection individuelles sont un bon complément.
En cas de grippe, les antiviraux sont plus efficaces s’ ils sont administrés durant les 48 premières heures.
1. Lang PO, Aspinall R. Vaccination in the elderly: what can be recommended? Drugs Aging 2014;31:581-99
2. McElhaney JE et al. The role of vaccination in successful independent ageing. Eur Geriatr Med 2016;7:171-5
3. Lang PO et al. Effectiveness of influenza vaccine in aging and older adults: comprehensive analysis of the evidence. Clin Interv Aging 2012;7:55-64
4. Demicheli V et al. Vaccines for preventing influenza in the elderly. Cochrane Database Syst Rev 2018, 2:CD004876. doi: 004810.001002/14651858.CD14004876.pub14651854
5. Demicheli V et al. Vaccines for preventing influenza in healthy adults. Cochrane Database Syst Rev 2018;2:CD001269. doi: 001210.001002/14651858.CD14001269.pub14651856
6. Jefferson T et al. Vaccines for preventing influenza in healthy children. Cochrane Database Syst Rev 2018;2:CD004879. doi: 004810.001002/14651858.CD14004879.pub14651855
7. Bartoszko JJ et al. Does consecutive influenza vaccination reduce protection against influenza: A systematic review and meta-analysis. Vaccine 2018;36:3434-44
8. Lang PO et al. Prior contacts with the 2000-2003 seasonal vaccines extends the 2009 pandemic A/H1N1 vaccine-specific immune protection to non-humoral compartments. Eur Geriatr Med 2014;5:136-8
9. Aspinall R LP. The Avalanche is Coming … And Just Now It’ s Starting to Snow. Front Immunol 2013;Jun 25;4:165
10. Office fédéral de la santé publique (OFSP): Grippe saisonnière 2017/2018 : réduire le risque de maladie pour soi et ses proches. OFSP – Bulletin 2017, 41:10-13
11. Lang PO et al. Influenza vaccination in the face of immune exhaustion: is herd immunity effective for protecting the elderly? Influenza Res Treat 2011;2011:419216
12. Imai C et al. A systematic review and meta-analysis of the direct epidemiological and economic effects of seasonal influenza vaccination on healthcare workers. PLoS One 2018;13:e0198685. doi: 0198610.0191371/journal.pone.0198685. eCollection 0192018
13. Seki M et al. Association of influenza with severe pneumonia/empyema in the community, hospital, and healthcare-associated setting. Respir Med Case Rep 2016;19:1-4
14. Mauskopf J et al. The burden of influenza complications in different high-risk groups: a targeted literature review. J Med Econ 2013;16:264-77
15. Lang PO. Pneumonies communautaires: ne pas oublier la grippe ! la gazette médicale 2016;6:36-7
16. Alimi Y et al. Systematic review of respiratory viral pathogens identified in adults with community-acquired pneumonia in Europe. J Clin Virol 2017;95:26-35
17. Baratali L, Lang PO. [Pneumococcal infections: Appraisal and perspectives in terms of adult vaccination]. Presse Med 2015;44:1155-61
18. Yin M et al. Effectiveness and safety of dual influenza and pneumococcal vaccination versus separate administration or no vaccination in older adults: a meta-analysis. Expert Rev Vaccines 2018;17:653-63
19. Lang PO. Vaccination antigrippe et vaccination antipneumococcique. Un outil de prévention cardiovasculaire ? la gazette médicale 2015;5:12-4
20. Offeddu V et al. Effectiveness of Masks and Respirators Against Respiratory Infections in Healthcare Workers: A Systematic Review and Meta-Analysis. Clin Infect Dis 2017;65:1934-42
21. MacIntyre CR, Chughtai AA: Facemasks for the prevention of infection in healthcare and community settings. BMJ 2015;350:h694
22. Prévention de la grippe et des infections respiratoires virales saisonnières [https://www.hcsp.fr/explore.cgi/avisrapportsdomaine?clefr=521]
23. Aspinall R, Lang PO. Vaccine responsiveness in the elderly: best practice for the clinic. Expert Rev Vaccines 2014;13:885-94
24. Aspinall R, Lang PO. Vaccination choices for older people, looking beyond age specific approaches. Expert Rev Vaccines 2018;17:23-30
25. Jefferson T et al. Vaccines for preventing influenza in the elderly. Cochrane Database Syst Rev 2010;Feb 17:CD004876
26. Grijalva CG et al. Association Between Hospitalization With Community-Acquired Laboratory-Confirmed Influenza Pneumonia and Prior Receipt of Influenza Vaccination. JAMA 2015;314:1488-97
27. Lang PO, Aspinall R. Vitamin D status and the host resistance to infections: What it is currently (not) understood. Clin Ther 2017;39:930-945
28. Ginde AA et al. High-dose monthly Vitamin D for prevention of acute respiratory infection in older long-term care residents: A randomized clinical trial. J Am Geriatr Soc 2017;65:496-503
29. Lang PO, Aspinall R. Can we translate vitamin D immunomodulating effect on innate and adaptive immunity to vaccine response? Nutrients 2015;7:2044-60
30. Yen HL. Current and novel antiviral strategies for influenza infection. Curr Opin Virol 2016;18:126-34
31. Organisation Mondiale de la Santé (OMS): Composition recommandée des vaccins antigrippaux pour la saison grippale 2014-2015 dans l’ hémisphère Nord. Wkly Epidemiol Rec 2014, 89:93-104
Au cours des 20 à 25 dernières années, les calculs rénaux sont devenus une maladie répandue en raison de changements dans le mode de vie et les habitudes alimentaires. La prévalence de la maladie a presque doublé pendant cette période. Les causes de la formation du calcul urinaire ne sont, en fin de compte, pas clairement élucidées. Cependant, les facteurs nutritionnels tels que la haute consommation de viande, les composants alimentaires riches en acide et la consommation excessive d’ alcool jouent un rôle important aux côtés des facteurs génétiques, de l’ apport en liquides et des conditions climatiques (1-3).
Le diagnostic et le traitement des calculs urinaires sont de plus en plus réguliers chez les médecins généralistes. L’ article suivant donne un aperçu du diagnostic, de la thérapie et de la métaphylaxie moderne de la lithiase rénale.
Diagnostic de calculs urinaires
En plus de l’ anamnèse et de l’ examen physique, l’ examen urinaire/ l’ analyse de laboratoire et l’ imagerie diagnostique sont d’ une importance fondamentale pour confirmer le diagnostic présumé d’ un calcul urinaire. En effet, en plus de la taille et de la localisation des calculs, l’ imagerie fournit des informations supplémentaires sur les pathologies associées ainsi que sur la fonction rénale (1, 2). Grâce à la disponibilité généralisée de l’ échographie et de la tomodensitométrie (TDM), ces procédures ont remplacé l’ ancienne procédure standard du pyélogramme intraveineux. En effet, cette dernière était d’ une grande précision.
Examen urinaire et laboratoire
L’ analyse d’ urine est une composante importante du diagnostic initial. La présence d’ une microhématurie peut confirmer le diagnostic présumé de néphropathie. La détection simultanée de bactéries combinée avec une augmentation de la CRP et des leucocytes peut être le signe d’ une infection compliquée des voies urinaires. En raison du danger d’ une stase urinaire septique menaçante et vitale, une clarification rapide et, si nécessaire, un drainage du rein est indiqué dans ces cas. La simple administration d’ une antibiothérapie ne suffit pas. Un test par culture d’ urine avec antibiogramme et de plus amples examens d’ imagerie diagnostique sont requis.
Échographie
Dans les cas aigus, l’ échographie est l’ imagerie de premier choix du praticien (1, 2). Un calcul rénal ou urétéral est représenté comme structure hyperéchogène avec annulation du son dorsal. Particulièrement dans le cas des calculs urétéraux, le calcul ne peut pas être détecté directement. De plus, le seul signe de l’ obstruction du drainage (4) est une dilatation du système pyélocaliciel du rein. En l’ absence de détection directe ou indirecte de calculs dans l’ échographie, il n’ est pas possible d’ exclure de manière fiable la présence d’ un calcul urétéral. En cas de suspicion clinique d’ un calcul urétéral, il faut procéder à une clarification supplémentaire au moyen d’ une TDM native.
Tomodensitométrie
En raison de sa bonne disponibilité, de son faible coût, de sa haute sensibilité et de sa spécificité, la TDM native est aujourd’ hui le Gold standard en matière de diagnostic de la lithiase urinaire. Elle donne des informations détaillées sur l’ anatomie des voies urinaires, les pathologies associées, le degré de dilatation ainsi que la localisation et la densité des calculs. Tous ces paramètres sont importants pour la planification ultérieure du traitement et devraient être inclus dans les recommandations du traitement (5-7). L’ imagerie par résonance magnétique (IRM) ne joue aucun rôle dans le diagnostic des calculs car ces derniers ne sont pas visibles par cette méthode.
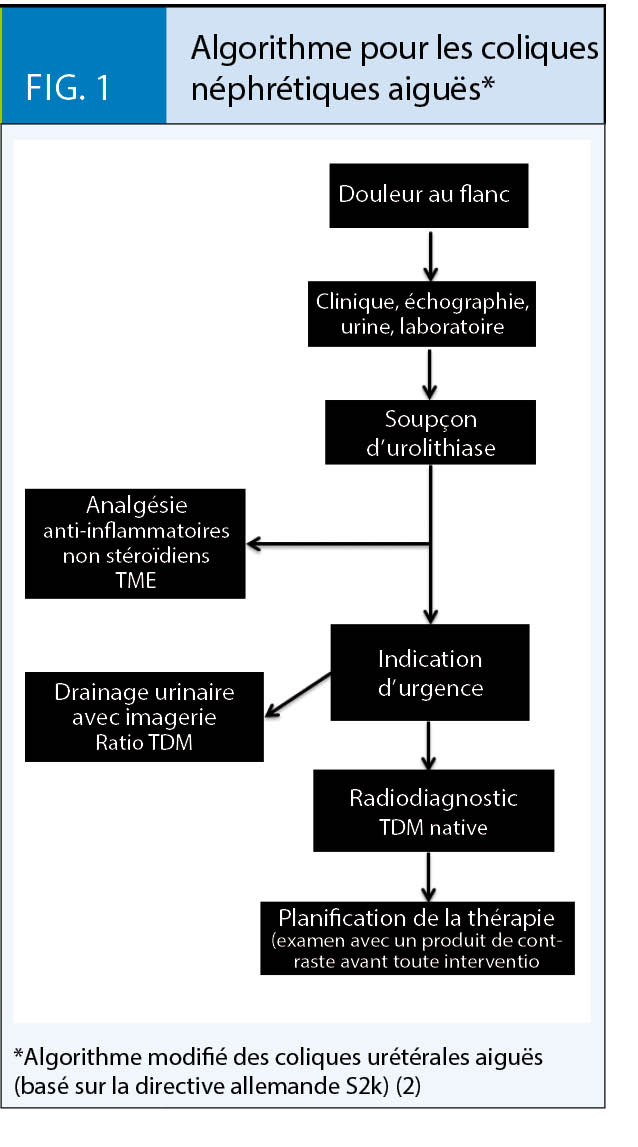
Thérapie conservatrice et interventionnelle des calculs
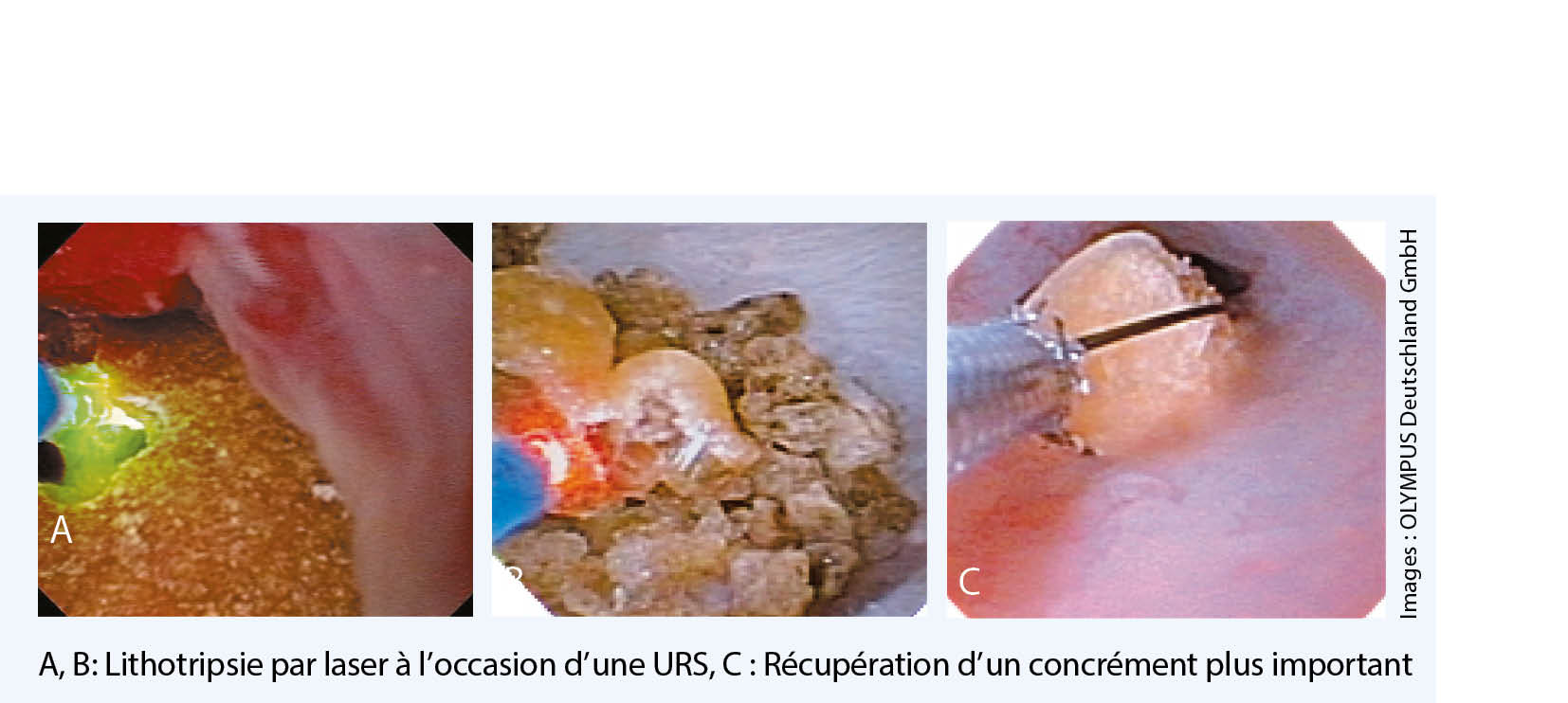
La thérapie des calculs rénaux doit toujours être une recommandation individuelle adaptée à la situation (fig. 1). Elle est basée sur l’ analyse clinique, l’intensité de la douleur, la localisation ainsi que la taille des calculs et des pathologies associées. De petits calculs urétéraux peuvent se détacher spontanément et ne nécessitent souvent pas d’ intervention (8). L’ introduction dans les années 80 de la lithotripsie par ondes de choc extracorporelles (LOCE) a révolutionné + la thérapie des calculs urinaires en brisant les calculs sans les toucher. Pendant des années, cette thérapie a été considérée comme le Gold standard et a été utilisée principalement pour toutes les maladies lithiasiques urinaires. De nouveaux développements et miniaturisations des lithotripteurs ont permis de réduire ainsi que de simplifier l’ utilisation technique de ces dispositifs par rapport à la première génération de la « baignoire à calculs ». Cependant, l’ efficacité a plutôt diminué en raison de ces changements techniques. D’ autres avancées techniques de ces dernières années ont, à nouveau, conduit à un changement de paradigme dans le traitement des calculs urinaires. Le développement d’ endoscopes plus fins et plus flexibles permet aujourd’ hui d’ effectuer une endoscopie à faible risque de l’ ensemble du tractus urinaire. En outre, elle représente, en combinaison avec la lithotripsie au laser, une procédure standard dans le traitement des calculs rénaux et urétéraux. Toutefois, l’ urétéro-rénoscopie (URS) a des limites pour des charges de calculs très élevées dans le rein. Dans ces cas, l’ ablation minimalement invasive des calculs parvoie endoscopique percutanée, la néphrolitholapaxie percutanée (NLPC), est très importante (9-11).
Thérapie conservatrice
Les petits calculs urétéraux (< 5 mm) ne nécessitent souvent pas de thérapie interventionnelle. Des études ont montré que jusqu’ à 95 % de ces calculs se détachent spontanément. Dans le cas de concréments plus importants (> 5 mm), le taux d’ élimination spontanée diminue considérablement. Les patients suivant un traitement conservateur doivent faire l’ objet d’ une surveillance étroite. L’efficacité de la thérapie médicamenteuse explosive (TME) avec des alpha-bloquants et des analgésiques n’ est finalement pas claire. Cependant, nous pouvons tout de même soutenir une élimination spontanée (12, 13). Une semaine plus tard, une réévaluation échographique devrait avoir lieu. Si le traitement est infructueux et/ou douloureux à cause de longues phases d’ expulsion, un traitement interventionnel peut être indiqué.
Thérapie interventionnelle
En plus de la LOCE, l’ URS est actuellement le traitement de choix pour les calculs rénaux et urétéraux (fig. 2). Grâce à cette procédure, les taux d’ élimination des calculs sont très élevés. De plus, dans les mains du praticien expérimenté, les risques et effets secondaires sont faibles (14). Le taux d’ élimination élevé après un seul traitement est l’ avantage décisif de l’ URS par rapport à la LOCE. Dans le cadre des soins d’ urgence, un cathéter Pigtail est généralement inséré dans l’ uretère. De cette manière, l’ uretère se dilate en une semaine et l’ URS peut être effectuée avec moins de risques ainsi qu’ avec un taux d’ élimination des calculs plus élevé. L’ urine doit tout de même être tamisée puisque même un cathéter Pigtail couché peut entraîner une élimination spontanée de calculs.
La NLPC est également utilisée aujourd’ hui pour des charges importantes de calculs avec épanchement partiel ou complet dans le bassinet rénal s’ il y a des calculs situés dans les diverticules du calice rénal ou bien après une URS non réussie. De nos jours, les opérations ouvertes pour éliminer les calculs sont très rares.
Métaphylaxie
En raison de la forte probabilité de récidive des calculs urinaires, la cause de l’ urolithiase doit être clarifiée. L’ analyse des calculs urinaires est ici d’ une importance primordiale. Des éclaircissements supplémentaires devraient suivre en fonction des risques. En particulier, les patients présentant les caractéristiques suivantes sont considérés comme étant à risque élevé et devraient être soumis à un examen spécifique de métaphylaxie des calculs urinaires par un spécialiste :
Récidives fréquentes (plus de 3 épisodes de calculs en 3 ans)
Enfants et adolescents
Formation de calculs déterminée génétiquement (cystinurie, hyperoxalurie primaire, acidose des tubules rénaux, xanthinurie)
Hyperparathyroïdie
Maladie gastro-intestinale (maladie de Crohn, colite ulcéreuse, statut après chirurgie bariatrique)
Zentrum für Urologie Zürich, Klinik Hirslanden
Witellikerstrasse 40
8032 Zürich
stephan.bauer@hirslanden.ch
L’ auteur a déclaré aucun conflit d’ intérêt en relation avec cet article.
L’ échographie est le premier choix pour le diagnostic primaire ainsi que pour le suivi. La tomodensitométrie à faible dose est utilisée pour approfondir le diagnostic dans les situations aiguës ainsi que pour la planification du traitement.
En cas de coliques néphrétiques aiguës, une analgésie avec des anti-inflammatoires non stéroïdiens est recommandée. En cas de persistance de la douleur et/ou d’ infection urinaire avec des signes accrus d’ inflammation, il faut procéder au drainage de l’ urine à l’ aide d’ un cathéter Pigtail.
Le traitement conservateur peut être effectué au moyen d’ analgésiques ainsi qu’ avec une thérapie médicamenteuse explosive. Un suivi étroit par échographie est indiqué.
L’ URS a presque remplacé la LOCE dans la thérapie interventionnelle des calculs urétéraux et rénaux. En fonction de la taille et de l’ emplacement des concréments, une thérapie individuelle peut être planifiée en utilisant des procédures mini-invasives. L’ URS a le taux le plus élevé d’ élimination complète des calculs après une seule intervention.
Des éclaircissements supplémentaires devraient prendre place en fonction de la classification des groupes à risque.
1. Knoll T et al (2016) S2k guidelines on diagnostics, therapy and metaphylaxis of urolithiasis (AWMF 043/025: Compendium. Urologe A 55(7):904-922
2. Türk C, Knoll T, Petrik A, Sarica K, Skolarikos A, Straub M, Seitz C (2016) EAU guidlines urolithiasis/.
3. Scales CD Jr. et al (2012) Prevalence of kidneystones in the United States. Eur Urol 62(1):160-165
4. Erwin BC, Carroll BA, Sommer FH (1985) Re: US in the evaluation of acute flank pain. Radiology 157 (2):554
5. Niall O et al (1999) A comparison of noncontrast computerized tomography with excretory urography in the assessment of acute flank pain. J Urol 161(2):543-537
6. Muller M et al (1998) The average dose-are product at intravenous urography in 205 adults. Br J Radiol 71(842):210-212
7. Yilmaz S et al (1998) Renal colic: comparison of spiral CT, US and IVU in the detection of ureteral calculi. Eur RAdiol 8(2):212-217
8. Miller OF, Kane CJ (1999) Time to stone passage for observed ureteral calculi: a guide for patient education. J Urol 162:688
9. Tiselius HG (2008) How efficient is extracorporeal shockwave lithotripsy with modern lithotripters for removal of ureteral stones? J Endourol 22(2):249-255
Die pädiatrische AML ist seltener aber aggressiver als die ALL. Heutzutage können ca. 60% der Patienten geheilt werden. Dieser Erfolg konnte erreicht werden, einerseits durch die Intensivierung der konventionellen Chemotherapie, eine umfassende supportive Therapie und eine sorgfältige Behandlung von Komplikationen und Rezidiven, andererseits durch eine zunehmend individualisierte prognostische Einordnung zum optimalen Einsatz verfügbarer Therapieoptionen.
La leucémie myéloïde aiguë pédiatrique est plus rare mais plus agressive que la leucémie lymphoblastique aigüe (LLA). Aujourd’ hui, environ 60 % des patients peuvent être guéris. Ce succès a été obtenu d’ une part par l’ intensification de la chimiothérapie conventionnelle, une thérapie de soutien complète et un traitement soigneux des complications et des récidives et, d’ autre part, par une classification pronostique de plus en plus individualisée pour une utilisation optimale des options thérapeutiques disponibles.
Epidemiologie
Die akute myeloische Leukämie (AML) gilt allgemein als Erkrankung des älteren Menschen. Bei jungen Leuten unter 20 Jahren hat sie eine Inzidenz von etwa 0,7 / 100.000 / Jahr (1). Wie bei den Erwachsenen wird zwischen primären und sekundären AML unterschieden. Die primären (oder «de novo») AML sind beim Kind deutlich häufiger und zeigen sich als akutes Krankheitsbild. Sekundäre AML entwickeln sich auf der Basis eines myelodysplastischen Syndroms (MDS) oder im Verlauf einer angeborenen (Fanconi Anämie, schwere kongenitale Neutropenie, Shwachman-Diamond Syndrom, Dyskeratosis congenita, u.a.) oder erworbenen aplastischen Anämie. Kinder mit Trisomie 21 haben ein 15-fach erhöhtes Risiko an Leukämie zu erkranken und entwickeln typischerweise ein transientes myeloproliferatives Syndrom als Neugeborene oder später das Vollbild einer AML, i.d.R. einer akuten megakaryozytischen Leukämie (AMKL-DS) (2).
Therapieinduzierte AML (t-AML) oder MDS (t-MDS) entstehen klassischerweise als Zweitmalignome einige Jahre nach Einsatz gewisser Zytostatika, die bekanntermassen kanzerogen wirken, insbesondere Anthrazykline und Epipodophyllotoxine (FAB M4 oder M5 Morphologie, typischerweise mit MLL-Rearrangement 11q23), Alkylantien (häufig mit Monosomie 7 oder Deletion 5q-) oder nach Radiotherapie.
Pathologie
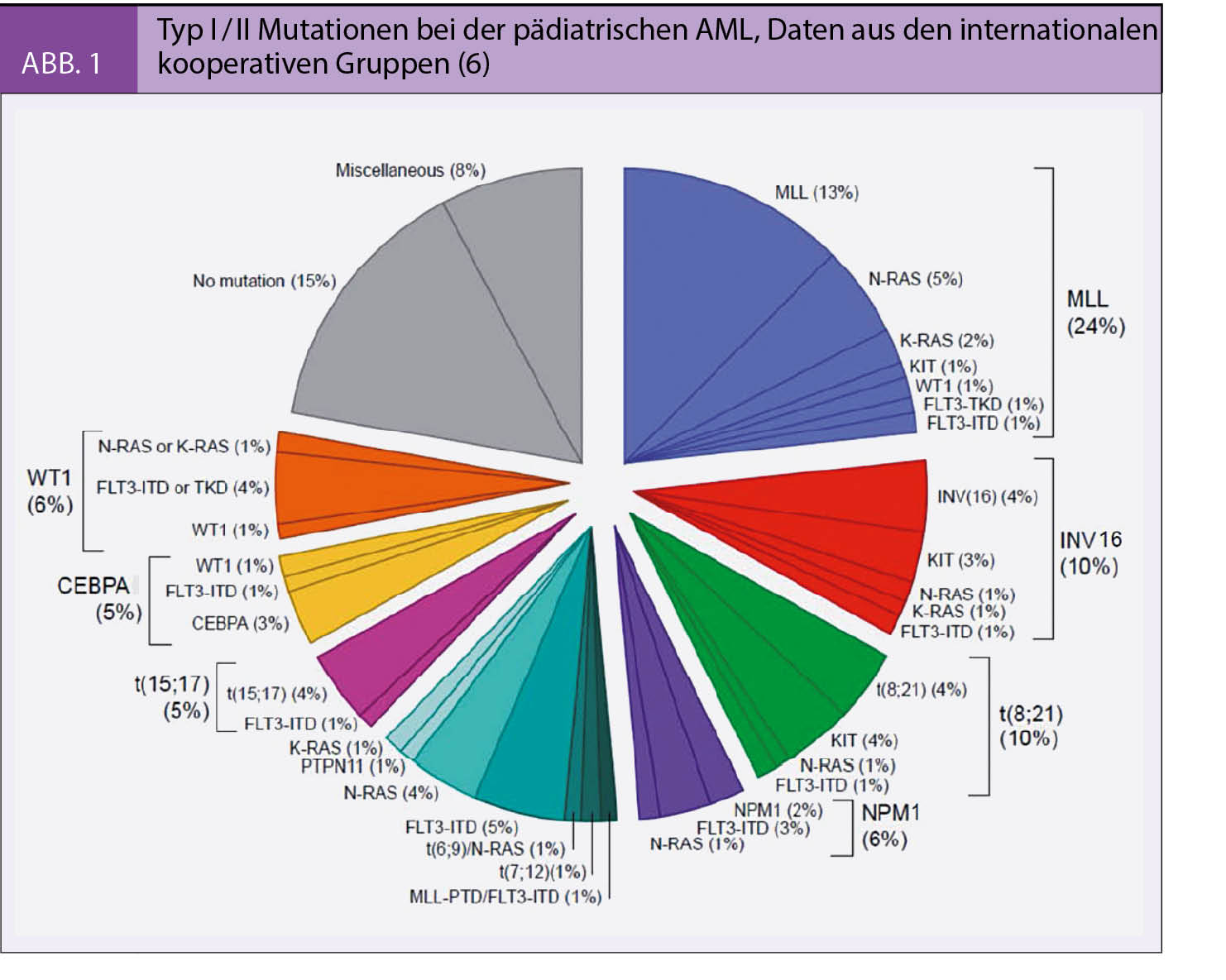
Bei der AML kommt es in frühen Progenitoren der Myelopoese oder in hämatopoetischen Stammzellen zur malignen Entartung. Zum Auftreten der offenen Leukämie sind wahrscheinlich mindestens zwei genetische Ereignisse in einem Zellklon erforderlich, die auch in grösserem zeitlichem Abstand auftreten können (3). Zudem spielen weitere Faktoren wie vulnerable Phasen der Immunentwicklung oder Veränderungen des Knochenmark-Mikroenvironments eine Rolle (4). Es gilt als gesichert, dass eine Differenzierungshierachie vorliegt, das heisst, dass sich der Grossteil der leukämischen Blasten aus der originären leukämischen Stammzelle (Stammzellklon) entwickelt (5). Bei der Mehrzahl der AML können leukämieassozierte zyto- und molekulargenetische Veränderungen nachgewiesen und entsprechend der Einteilung von Gilliland et al. als Typ I oder Typ II Mutationen (Abb. 1) eingeordnet werden, die entweder isoliert oder kombiniert nachweisbar sind (6). In einer umfassenden europäischen Kollaboration zur AML bei Kindern und Jugendlichen konnten die Häufigkeit und prognostische Relevanz dieser Mutationen belegt werden (7, 8, 9). Heutzutage sind diese Erkenntnisse ans Licht der Entwicklung molekular wirkender Substanzen, die als individualisierte, rationale Therapieoptionen infrage kommen, von besonderer Bedeutung.
Therapie
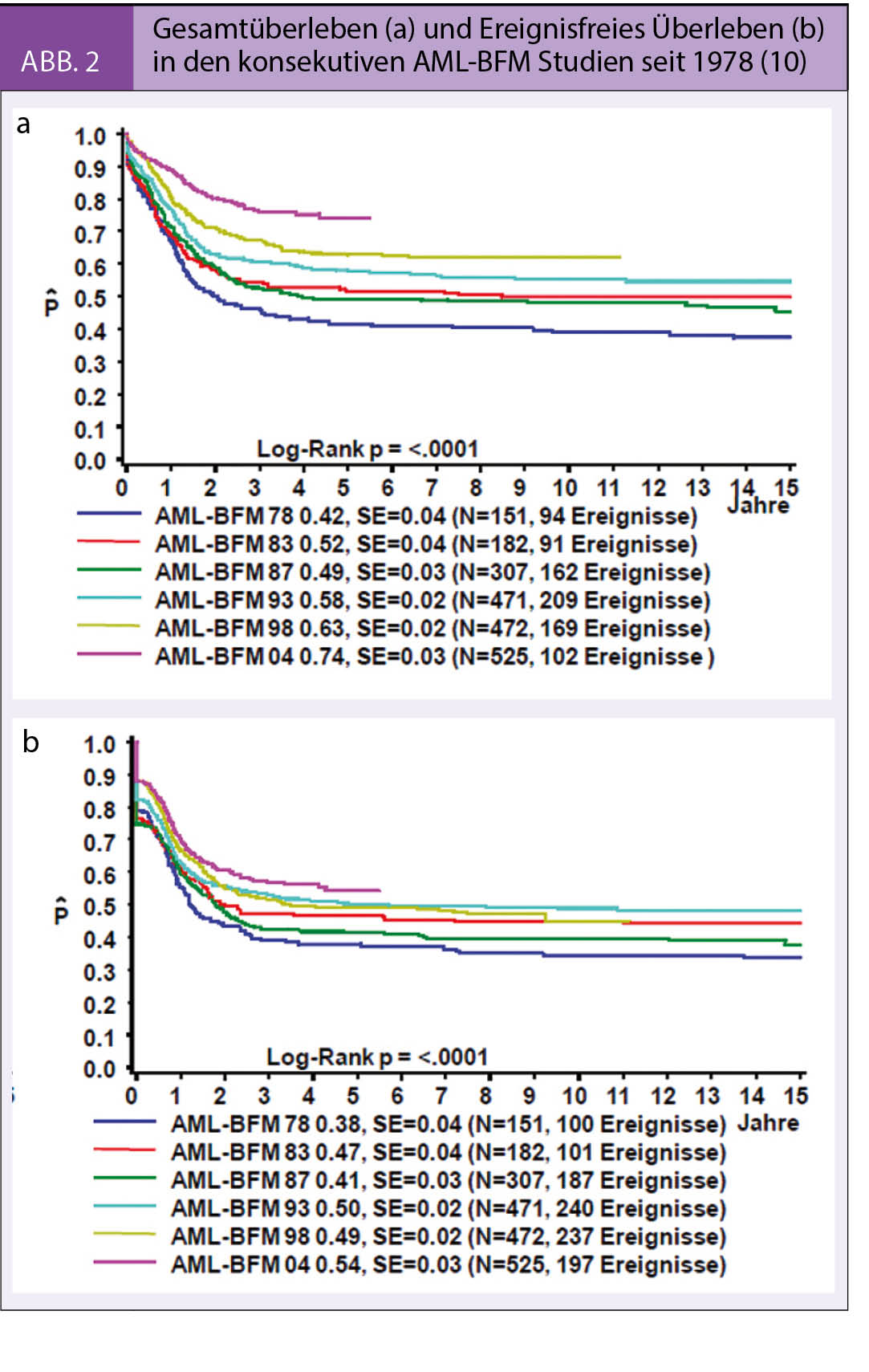
Die Prognose der pädiatrischen AML hat sich im Laufe der letzten 40 Jahre dank der Entwicklung und Umsetzung internationaler Therapieoptimierungsstudien (u. a. AML-BFM Gruppe in Europa, POG / COG in Amerika) kontinuierlich verbessert. Die 10-Jahres-Überlebensrate lag Ende der 70-er Jahre bei 42% und konnte bis heute auf 74% verbessert werden (Abb. 2) (10). Dieser Fortschritt wurde einerseits durch die Verbesserung der Kenntnisse der Molekularbiologie der AML mit anschliessender Verfeinerung der Therapiestratifizierung der Patienten, andererseits durch eine deutliche Intensivierung der Primärtherapie –insbesondere der Induktionstherapie – sowie durch verbesserte prophylaktische und supportive Massnahmen erreicht. Hinzu kommt eine konsequentere und verstärkte Behandlung der Rezidive (11). Weltweit wird aktuell die pädiatrische AML mit kurzen und intensiven Therapieblöcken behandelt, welche hauptsächlich aus Cytosinarabinosid (Ara-C) in verschiedenen Dosenintensitäten, Anthrazyklinen (Daunorubicin, liposomales Daunorubicin, Idarubicin und Mitoxantron) sowie Etoposid bestehen.
Wie in der ALL hat auch in der AML, neben den genetischen und biologischen Eigenschaften der Krankheit, das frühe Therapieansprechen einen sehr hohen prognostischen Stellenwert. Die Entwicklung hochsensitiver Methoden zur Erfassung der sogenannten minimal residuellen Erkrankung (minimal residual disease, MRD) und die entsprechende Anpassung der Therapieintensität, insbesondere früh in der Behandlung, haben somit eine zentrale Rolle in der Besserung der Resultate gespielt. Zur MRD-Bestimmung kommen prinzipiell methodisch die Morphologie, Immunphänotypisierung, Monitoring von genetischen Aberrationen (FISH) oder Fusionsgenen (qPCR), Monitoring ausgewählter Genexpressionen und die Nachverfolgung von klonspezifischen Mutationen (qPCR) infrage.
Die verschiedenen diagnostischen Methoden unterscheiden sich insbesondere hinsichtlich der Sensitivität (Morphologie <FISH < Immunphänotypisierung < qPCR) und Spezifität (Mutationen > Fusionsgene > FISH > Immunphänotypisierung > Morphologie) (12). Wichtige Aspekte der Anwendung dieser Technologien sind darüber hinaus die Zielsetzung und der Zeitpunkt innerhalb der einzelnen Therapiephasen. Für die Bestimmung des Therapieansprechens können prinzipiell alle Methoden herangezogen werden, da es in erster Linie auf die Kinetik ankommt. Für das spätere Monitoring, mit der Zielsetzung ein drohendes Rezidiv (molekulares Rezidiv) frühzeitig zu erkennen, kommen nur sehr spezifische und sensitive Methoden infrage.
Die zunehmenden Verbesserungen der diagnostischen Methoden (multi-color-Flowcytometry) und die Identifikation neuer Mutationen (FLT3-ITD/TDK, NPM1, c-kit, ras, CEBPΑ etc.) könnten die Aussagefähigkeit der MRD-Diagnostik entscheidend verbessern und eine noch geeignetere Therapiestratifizierung sowohl im Sinne der Therapiereduktion bei gutem Ansprechen als auch der Intensivierung bei ungünstigem Ansprechen ermöglichen (13).
Dieses bekommt einen umso höheren Stellenwert, als dass durch die Einführung einer Vielzahl neuer Inhibitoren/molekular wirksamer Substanzen, zusätzliche Werkzeuge zur Bestimmung des Therapieansprechens erforderlich werden.
Rolle der ZNS-Bestrahlung
Ein initialer ZNS-Befall der AML ist in ca. 5-15% der Kinder vorhanden. Verschiedene Studien haben gezeigt, dass eine intensive, wöchentliche intrathekale Therapie bis zur Klärung des Liquors genau so wirksam ist wie die ZNS-Bestrahlung. Dies gilt auch bei der Behandlung der meisten Chlorome. Auch die prophylaktische ZNS-Behandlung mit systemisch und intrathekal applizierter Chemotherapie hat sich gegen die Bestrahlung durchgesetzt. In der aktuellen AML-BFM 2012 Studie wird auf die prophylaktische ZNS-Bestrahlung verzichtet. Es erfolgen je nach Therapiearm 9 bzw. 11 intrathekale Tripletherapien (Prednison, Methotrexat und Cytarabin), ausser bei gleichzeitiger Applikation von intravenösem hochdosiertem Cytarabin, um unnötige Neurotoxizität zu vermeiden. Hier besteht die intrathekale Therapie aus einer Monotherapie mit Cytarabin.
Stellenwert der Erhaltungstherapie
International ist die Erhaltungstherapie umstritten. Aus Studien mit Erwachsenen ist bekannt, dass die Ergebnisse bei weniger intensiver Therapie mit Erhaltungstherapie für einen Teil der Patienten von Vorteil sind (14), jedoch bei Einsatz einer intensiven Anfangstherapie keinen Vorteil bringen (15).
Bei Kindern wurde in der französischen LAME-Studie 89/91 gezeigt, dass die Ergebnisse mit und ohne Erhaltungstherapie für das erkrankungsfreie Überleben (DFS) im gleichen Bereich liegen, während die Wahrscheinlichkeit des Gesamtüberlebens (OS) sogar besser war, wenn keine Erhaltungstherapie durchgeführt wurde – begründet durch eine höhere Salvage-Rate nach Rezidiven ohne vorherige Erhaltungstherapie (16). Ebenso zeigte die amerikanische CCG-Studie 213, dass die Erhaltungstherapie nach einer Intensivierung mit Hochdosis-Cytarabin nicht notwendig war. Andererseits spielte sie eine Rolle im Zweig mit Standardintensität in der Induktion (17). Aufgrund der Heterogenität der AML und der unterschiedlichen Proliferationskinetik ist es auch denkbar, dass nur für bestimmte Subtypen der AML eine Erhaltungstherapie von Vorteil sein kann.
Stellenwert der allogenen Stammzelltransplantation
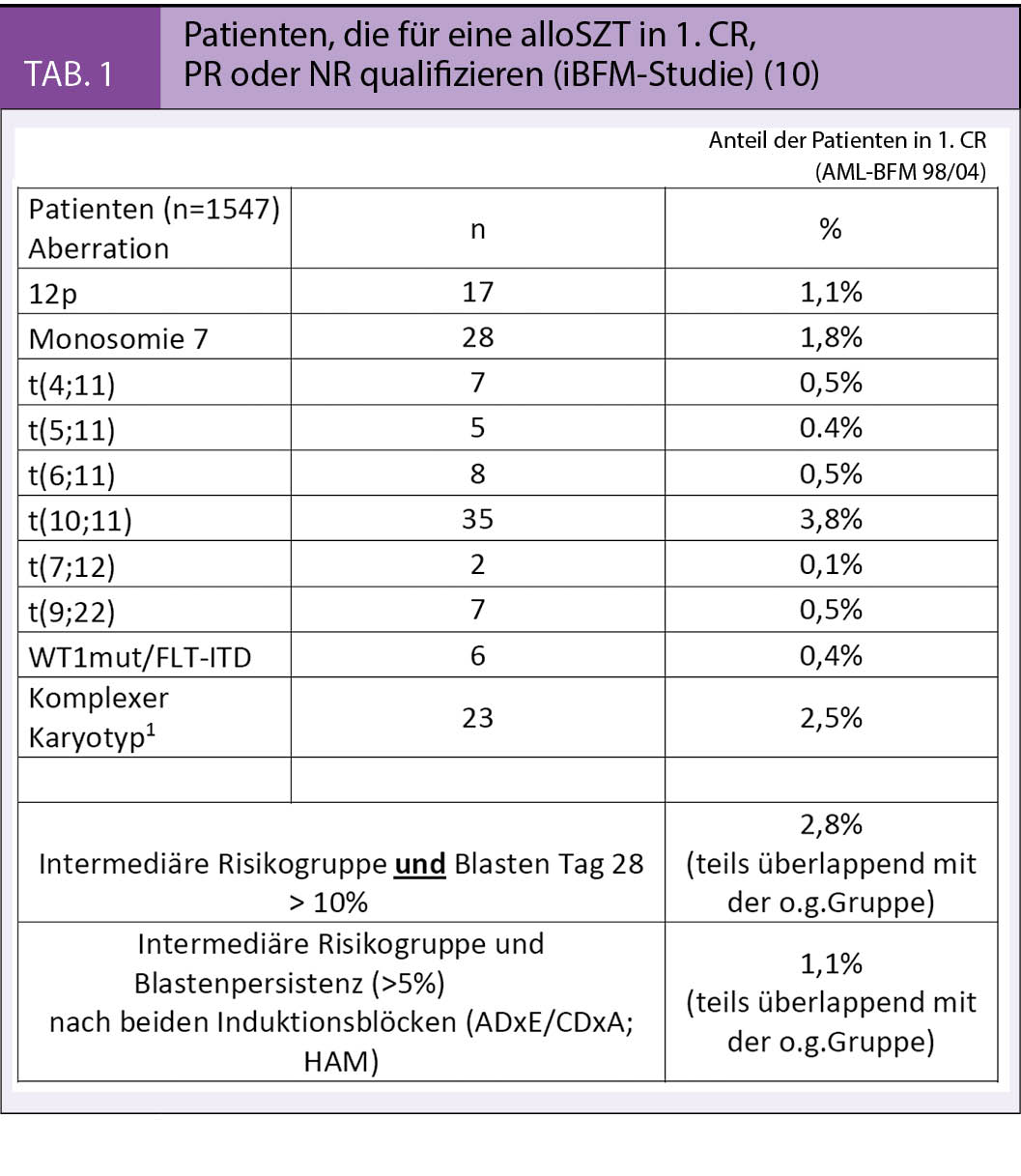
Der Stellenwert der allogenen Stammzelltransplantation (alloSZT) in erster kompletter Remission (1. CR) wird nach wie vor diskutiert. Die prospektive Analyse der alloSZT von einem Geschwisterspender für die Hochrisikogruppe der AML-BFM 98 Studie konnte keinen signifikanten Vorteil belegen. Dieses deckt sich mit den Ergebnissen der britischen MRC-Studie und der skandinavischen NOPHO Studie. Andererseits ergab eine Metaanalyse bei Kindern und Jugendlichen einen Vorteil der alloSZT in 1. CR (18). Die Aufarbeitung der verschiedenen Studien weist daraufhin, dass die Ergebnisse der alloSCT in den verschiedenen Gruppen ähnlich sind in Bezug auf das Rezidivrisiko, die Morbidität und das Überleben. Deutlichere Unterschiede treten eher beim Vergleich der jeweiligen Chemotherapiearme auf. Hinzu kommt, dass ein möglicher Vorteil der SZT auch in den unterschiedlichen Risikogruppen sehr unterschiedlich ist (19).
Die Tabelle 1 stellt die aktuellen Indikationen der iBFM AML Gruppe für eine alloSZT sowohl vom Geschwister- als auch vom passenden Fremdspender in der Frontline Therapie dar. Alle prognostisch ungünstigen molekular-biologischen Subtypen sind dabei vertreten, sowie die Patienten mit fehlendem Ansprechen auf die Induktionstherapie (sog. Non-Responders, ca. 4% der Hochrisikopatienten) für welche die alloSZT die einzige kurative Option bietet.
Unumstritten ist die SZT jedoch im Rezidiv Fall.
Neue Therapien
Trotz allen Fortschritten bleibt die Prognose der pädiatrischen AML deutlich unter jener der ALL. Patienten mit refraktärer AML (non-responders), diejenigen mit Rezidiv nach allogener Stammzelltransplantation aber auch einige Patienten mit bestimmten sehr Hoch-Risiko Mutationen haben alle noch sehr eingeschränkte Überlebenschancen. Deshalb sind neue, wenn möglich gezielte, Therapieeinsätze notwendig. Dank der oben erwähnten Fortschritte in der molekular-biologischen Charakterisierung der AML konnten eine Vielzahl neuer Mutationen (Typ I / II, Abb. 1) nachgewiesen werden, die sowohl bei der Leukä-mogenese als auch für Proliferation, Differenzierungsgrad oder Phänotyp relevant sind, aber auch therapeutisch genutzt werden können. Es wurden spezifische Substanzen (Tyrosin-Kinase Inhibitoren, «small molecules» etc.) entwickelt, die einzelne Signalkaskaden blockieren können. Trotz der meist guten Effektivität in vitro, ergaben die klinischen Studien als Monotherapien in der Regel eher begrenzte Behandlungserfolge. In Kombination mit konventionellen Chemotherapien ergeben sich allerdings zunehmend Hinweise, dass bei einem gezielten Einsatz dieser Substanzen, die Ansprechraten von AML-Patienten verbessert werden können.
Sorafenib (als Beispiel einer gezielten Therapie)
Sorafenib ist ein Arzneimittel aus der Gruppe der Tyrosin-Kinase-Inhibitoren (TKI) mit Wirkung auf multiple Tyrosinkinasen (multi-TKI) und hat somit mehrere Angriffspunkte: i) Inhibition der Rezeptor-Tyrosinkinasen (FLT-3,c-kit, VEGFR-2, VEGFR-3, PDGFR-b) mit Blockade der entsprechenden Signalkaskaden und reduzierter Tumor-Angiogenese; ii) Inhibition der Serin/Threonin-Kinasen (Raf-Kinasen: CRAF, BRAF, V600EBRAF) mit Hemmung der Raf-Signalkaskade. In beiden Fällen führt Sorafenib zu einer verminderten Zellteilung und Proliferation. Vorläufige Ergebnisse in Fallberichten und Studien bei Erwachsenen zeigen, dass eine gezielte Therapie mit Sorafenib bei AML mit FLT3-ITD eine vielversprechende Option anbieten könnte (20). Weitere experimentelle Daten zeigen, dass Inhibitoren wie Sorafenib, die die FLT3-ITD spezifischer hemmen als andere multi-TKI (Lestaurtinib[CEP701], Midostaurin [PKC412]), effektiver für FLT3-ITD/TDK positive AML sein können und weniger Nebenwirkungen zeigen (21).
Das gegenwärtige Therapieprotokoll der iBFM Gruppe (International Relapsed AML 2010/01) wird die Wirkmechanismen dieses TKI bei Patienten mit einer FLT3-ITD berücksichtigen. Durch die sequenzielle Gabe von Sorafenib (3 Tage Intervall zwischen Sorafenib und Chemotherapiestart) wird die gleichzeitige Applikation mit der Chemotherapie vermieden und somit das Risiko einer möglichen Wirkminderung der Chemotherapie durch Änderung des Zellzyklus reduziert. Zudem werden die mehrfach in vitro nachgewiesenen schnellen aber transienten Resistenzentwicklungen vermieden, die höchstwahrscheinlich nicht auf einen resistenten Klon zurückzuführen sind.
Der Autor hat keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
Die pädiatrische AML ist eine seltene, molekularbiologisch sehr
heterogene Erkrankung
Die Behandlung beruht auf kurze, intensive und sehr myelotoxische Chemotherapieblöcke
Wie bei der ALL ist eine Stratifizierung der Patienten auf der Basis der Molekularbiologie und des frühen Ansprechens unentbehrlich
Der Stellenwert der alloSZT in der initialen Therapie ist umstritten, für bestimmte Subtypen der AML aber sicher relevant
Neue Therapien, insbesondere TKI, könnten die düstere Prognose bestimmter Patientengruppen verbessern
Messages à retenir
La leucémie myéloïde aiguë pédiatrique est une maladie rare, moléculairement très hétérogène
Le traitement repose sur des blocs de chimiothérapie courts, intensifs et très myélotoxiques
Comme pour la LLA, la stratification des patients sur la base de la biologie moléculaire et de la réponse précoce est indispensable
Le rôle de la transplantation de cellules souches allogéniques en traitement initial est controversé, mais certainement pertinent pour certains sous-types de leucémie myéloïde aiguë
De nouvelles thérapies, en particulier les ITK, pourraient améliorer le pronostic sombre de certains groupes de patients
1. Kaatsch P, Spix C. Jahresbericht / Annual Report 2005. Book. 2008.
2. Zwaan MC, Reinhardt D, Hitzler J, Vyas P. Acute leukemias in children with Down syndrome. Pediatr Clin North Am. 2008;55:53-70,x.
3. Greaves M. Childhood leukaemia. BMJ. 2002;324:283-287.
4. Rubnitz JE, Gibson B, Smith FO. Acute myeloid leukemia. Pediatr Clin North Am. 2008;55:21-51, ix.
5. Appelbaum FR, Rowe JM, Radich J, Dick JE. Acute myeloid leukemia. Hematology (Am Soc Hematol Educ Program ). 2001;:62-86.
6. Lane SW, Gilliland DG. Leukemia stem cells. Semin Cancer Biol. 2009.
7. Goemans BF, Zwaan CM, Miller M, Zimmermann M, Harlow A, Meshinchi S, Loonen AH, Hahlen K, Reinhardt D, Creutzig U, Kaspers GJ, Heinrich MC. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia.
8. Hollink IHIM, Zwaan CM, Zimmermann M, Arentsen-Peters TCJM, Pieters R, Cloos J, Kaspers GJL, de Graaf SSN, Harbott J, Creutzig U, Reinhardt D, Heuvel-Eibrink MM, Thiede C. Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia. 2009;23:262-270.
9. Zwaan CM, Meshinchi S, Radich JP, Veerman AJ, Huismans DR, Munske L, Podleschny M, Hahlen K, Pieters R, Zimmermann M, Reinhardt D, Harbott J, Creutzig U, Kaspers GJ, Griesinger F. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood. 2003;102:2387-2394.
10. Creutzig U, Zimmermann M, Ritter J, Reinhardt D, Hermann J, Henze G, Jurgens H, Kabisch H, Reiter A, Riehm H, Gadner H, Schellong G. Treatment strategies and long-term results in paediatric patients treated in four consecutive AML-BFM trials. Leukemia. 2005;19:2030-2042.
11. Sander A, Zimmermann M, Dworzak M, Fleischhack G, von Neuhoff C, Reinhardt D, Kaspers GJ, Creutzig U. Consequent and intensified relapse therapy improved survival in pediatric AML: results of relapse treatment in 379 patients of three consecutive AML-BFM trials. Leukemia. 2010;24:1422-1428.
12. Reinhardt D, Langebrake C, Creutzig U, Vormoor J, Brune C, Thorwesten M, Ingiliz P, Hrusak O, Dworzak M, Griesinger F. Minimal residual disease in acute myeloid leukemia in children – standardization and evaluation of immunophenotyping in the AML-BFM-98 study. Klinische Padiatrie. 2002;214:179-187.
13. Kern W, Haferlach C, Haferlach T, Schnittger S. Monitoring of minimal residual disease in acute myeloid leukemia. Cancer. 2008;112:4-16.
14. Büchner T, Hiddemann W, Löffler H, Nowrousian MR, Maschmeyer G, Aul HC, Straif K, Hossfeld D, Heinecke A, for the AMLCG. Long-term results in adult AML: maintenance versus no maintenance, and double versus standard induction. Blood. 1990;74, Suppl. 1:105.
15. Mandelli F, Vegna ML, Avvisati G, et al. A randomized study of the efficacy of postconsolidation therapy in adult acute nonlymphocytic leukemia: a report of the Italian Cooperative Group GIMEMA. Ann Hematol. 1992;64:166-172.
16. Perel Y, Auvrignon A, Leblanc T, Vannier JP, Michel G, Nelken B, Gandemer V, Schmitt C, Lamagnere JP, de Lumley L, Bader Meunier B, Couillaud G, Schaison G, Landman-Parker J, Thuret I, Dalle JH, Baruchel A, Leverer, and for the Group LAME of the French Society of Pediatric Hematology and Immunology. Impact of addition of maintenance therapy to intensive induction and consolidation chemotherapy for childhood acute myeloblastic leukemia: results of a prospective randomized trial, LAME 89/91 [abstract]. J Clin Oncol. 2002;20:2774-2782.
17. Wells RJ, Woods WG, Lampkin BC, Nesbit ME, Lee JW, Buckley JD, Versteeg C, Hammond GD. Impact of high-dose cytarabine and asparaginase intensification on childhood acute myeloid leukemia: a report from the Children’s Cancer Group. J Clin Oncol. 1993;11:538-545.
18. Bleakley M, Lau L, Shaw PJ, Kaufman A. Bone marrow transplantation for paediatric AML in first remission: a systematic review and meta-analysis. Bone Marrow Transplant. 2002;29:843-852.
19. Horan J, Korones D. Intensive chemotherapy and bone marrow transplantation for children with acute myeloid leukemia. Blood. 2001;97:3672-3673.
20. Metzelder S, Wang Y, Wollmer E, Wanzel M, Teichler S, Chaturvedi A, Eilers M, Enghofer E, Neubauer A, Burchert A. Compassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: sustained regression before and after allogeneic stem cell transplantation. Blood. 2009;113:6567-6571.
21. Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, Grandin W, Lebwohl D, Wang Y, Cohen P, Fox EA, Neuberg D, Clark J, Gilliland DG, Griffin JD. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54-60.
MDS sind klonale Bluterkrankungen des älteren Menschen und werden vorwiegend bei Personen im Alter über 70 Jahre diagnostiziert. In europäischen Ländern beträgt die alters-standardisierte Inzidenzrate 2-3 pro 100’000 Patientenjahre mit einer zweifach höheren Inzidenz bei Männern als bei Frauen. Die einzige Ausnahme stellt dabei das MDS mit del(5q) dar, welche eine weibliche Prädominanz hat (1-4). Aufgrund der demographischen Alterung und der zunehmenden diagnostischen Möglichkeiten muss man in Zukunft davon ausgehen, dass die Entität der MDS zu einer der häufigsten hämatologischen Neoplasien aufsteigen wird, mit relevanter Auswirkungen auf die Gesundheitsversorgung (1).
Les MDS sont des maladies clonales du sang des personnes âgées qui sont principalement diagnostiquées chez les personnes de plus de 70 ans. Dans les pays européens, le taux d’incidence normalisé selon l’âge est de 2 à 3 pour 100 000 années-de patients, l’incidence étant deux fois plus élevée chez les hommes que chez les femmes. La seule exception est le MDS avec del (5q), qui a une prédominance féminine (1-4). En raison du vieillissement démographique et de l’augmentation des possibilités de diagnostic, le MDS devrait devenir l’une des néoplasies hématologiques les plus courantes à l’avenir, avec un impact important sur les soins de santé (1).
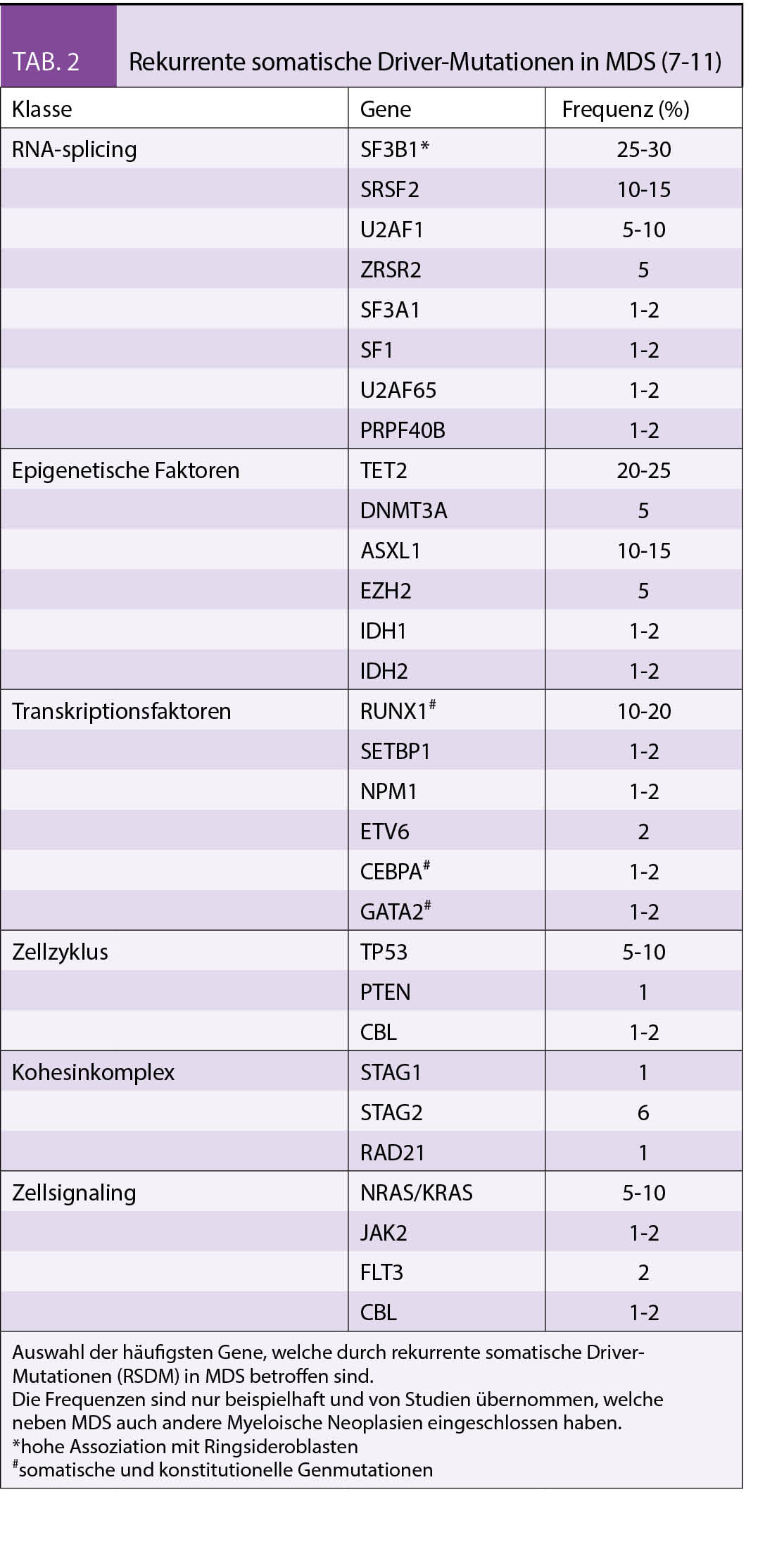
MDS sind heterogene Erkrankungen, die durch sequenzielle Ansammlung von genetischen Läsionen in den hämatopoetischen Stammzellen (HSC) verursacht werden (5). Die genetischen Läsionen, die bisher bei MDS identifiziert werden konnten, waren strukturelle Chromosomenaberrationen, die sich durch eine konventionelle Metaphasen-Zytogenetik oder Fluoreszenz-In-Situ Hybridisierung (FISH) nachweisen lassen. Die Analyseverfahren und dadurch auch das Verständnis genetischer Veränderungen in MDS und anderen myeloischen Neoplasien haben sich jedoch in den letzten Jahren rasant weiterentwickelt (6). Dank der Next Generation Sequencing (NGS) ist es nun möglich, rekurrente somatische Driver-Mutationen (RSDM) nachzuweisen. Diese RSDM treten in Genen mit folgenden Funktionen auf: RNA-Splicing, epigenetische Regulation, Transkriptionsfaktoren, Zellzyklus, Kohesinkomplex und Zellsignalling (7 - 11).
Diagnostik und Klassifizierung
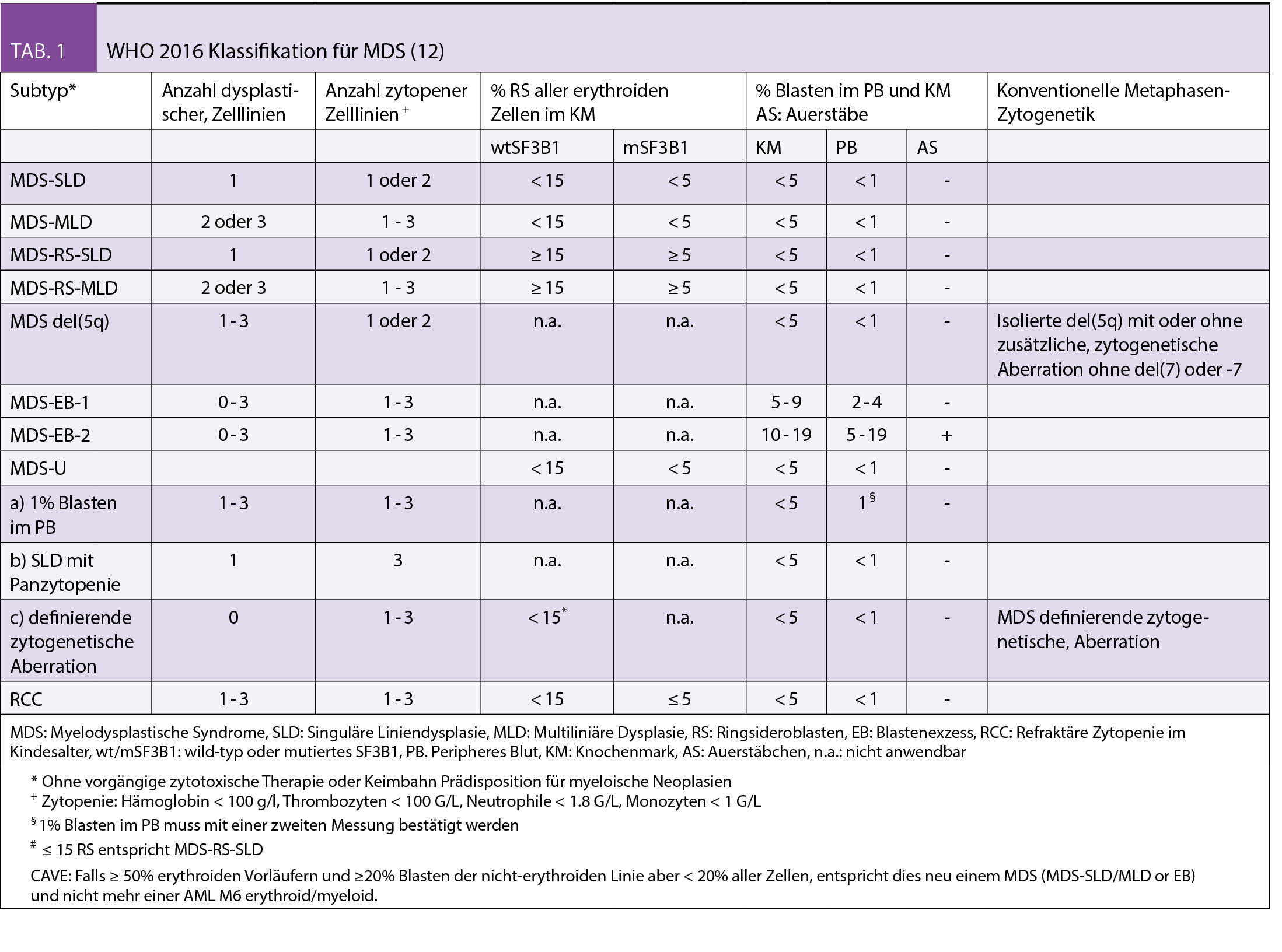
Die diagnostischen Empfehlungen bei erwachsenen Patienten mit vermutetem MDS wurden in den ELN-Empfehlungen 2013 zusammengefasst (7). Eine gründliche persönliche Anamnese (symptomatische Anämie, Infekte, Blutungen und insbesondere Expositionen gegenüber Chemo-/Radiotherapie, Pestizide, Insektizide und Lösungsmittel), Familienanamnese (Hinweise für hereditäre Prädisposition bei jüngeren MDS Patienten) sowie eine körperliche Untersuchung (Blutungszeichen, Organomegalien, Lymphadenopathien) stellen eine wichtige Grundlage dar. Zwingende Laboranalysen umfassen die morphologische Beurteilung des peripheren Blutes, des Knochenmarkaspirates/der Biopsie und eine zytogenetische Analyse. Zusätzlich empfohlene Untersuchungen umfassen die FISH (falls Zytogenetik nicht aussagekräftig) sowie die fluoreszenzaktivierte Zellsortierung (FACS). In speziellen Situationen werden zusätzlich auch molekulare Analysen (array-CGH, PCR und NGS) zum Nachweis von kryptischen Gendefekten und RSDMs empfohlen. Für die Klassifizierung der MDS werden die Anzahl von Zell-Linien, die von Zytopenie und Dysplasie betroffen sind, das Vorhandensein von Ringsideroblasten (RS) oder der Mutationen in SF3B1 (das mit RS assoziiert ist), Anzahl Blasten im peripheren Blut oder Knochenmark und das Vorliegen von speziellen MDS-definierenden zytogenetischen Anomalien (zB del 5q) berücksichtigt (Tab. 1) (12).
Zytopenien im Alter
Eine ungeklärte Anämie findet sich bei etwa 10 - 15% der Patienten im Alter von > 65 Jahren und es ist nicht immer einfach, reaktive von klonalen Zuständen abzugrenzen. Patienten mit einer Zytopenie, jedoch ohne ausreichende dysplastische Veränderungen oder MDS-definierende zytogenetische Veränderungen, werden als idiopathische Zytopenie unklarer Signifikanz (ICUS) bezeichnet (13). Bei diesen Patienten wird eine Verlaufskontrolle nach 3 - 6 Monaten und ggf. eine Wiederholung der Knochenmarksuntersuchung empfohlen (7). RSDM können mit einer altersabhängigen, erhöhten Häufigkeit bei älteren Patienten (10-20%) nachgewiesen werden. Diese Personen haben normale periphere Blutwerte oder nur eine leichte Zytopenie, welche aber die diagnostischen Kriterien für MDS nicht erfüllen. Diese Zustände werden als klonale Hämatopoiese mit indeterminiertem Potential (CHIP: normale periphere Blutwerte ohne RSDM) (14, 15) oder klonale Zytopenie unklarer Signifikanz (CCUS: Zytopenie mit RSDM) (16) bezeichnet. Die Anwendung der NGS ermöglicht neuerdings Patienten mit reaktiven und klonalen Zuständen zu unterschieden. Auf diese Weise lassen sich Patienten in frühen Stadien einer klonalen Hämatopoiese identifizieren, welche ein erhöhtes Risiko für die Entwicklung einer overten hämatologischen Neoplasie haben.
Allgemeine Überlegungen zum MDS Patientenmanagement
MDS sind sehr heterogene Erkrankungen mit einem sehr variablen natürlichen Verlauf von chronischen, asymptomatischen Zytopenien bis zu einem schnellen Fortschreiten in eine sekundäre AML. Zwei Drittel der Patienten mit MDS sterben an zytopenieassoziierten Komplikationen und ein Drittel erliegt der AML-Progression (17). Das Management basiert auf einer krankheits- und patientenassoziierten Risikostratifizierung. Eine exakte Diagnose und Risikostratifizierung sind für eine korrekte Behandlung daher entscheidend. Patienten mit niedrigem Risiko haben ein medianes Überleben von 3 bis 8 Jahren auf und sterben vorwiegend an zytopenieassoziierte Komplikationen (kardiovaskuläre Ereignisse, Infektionen und Blutungen). Die Ziele einer Behandlung von Patienten mit niedrigem Risiko liegen daher vor allem in der Verbesserung der Lebensqualität, Verringerung von zytopenieassoziierten Komplikationen und einem Hinauszögern eines Progresses in ein höhergradiges MDS (18, 19). Bei MDS Patienten mit hohem Risiko liegt hingegen die mediane Überlebenszeit nur bei 1 bis 3 Jahre und die AML-assoziierte Mortalität steht im Vordergrund. Bei diesen Patienten sollte die Behandlung darauf ausgerichtet sein, die Progression in eine AML hinauszuzögern und das Gesamtüberleben zu verbessern (20, 21).
Krankheitsassoziierte Risikofaktoren
Das Risiko für eine Progression in eine sekundäre AML und das Gesamtüberleben kann durch den International Prognostic Scoring System (IPSS) und den revidierten IPSS (IPSS-R) abgeschätzt werden) (22 - 24). Die Anzahl der Zelllinien, die von Zytopenien betroffen sind, wird in allen Prognosescoring-Systemen verwendet. Zudem sind Blastenzahl und Art der zytogenetischen Veränderungen relevant. Weniger als 5% der Blasten im Knochenmark, keine zirkulierenden Blasten im peripheren Blut, isolierte Anämie, Transfusionsunabhängigkeit und normaler Karyotyp oder günstige zytogenetische Veränderungen charakterisieren weniger fortgeschrittene MDS Formen. Im Gegensatz dazu wird ein fortgeschrittenes MDS durch > 5% Blasten im Knochenmark, Zytopenien mehrerer Zelllinien, komplexe zytogenetische oder andere ungünstige Veränderungen definiert.
Patientenassoziierte Risikofaktoren
Bei onkologischen Behandlungen älterer Patienten ist es stets wichtig, die Wirksamkeit und die Verträglichkeit einander gegenüber zu stellen. Karnofsky und Eastern Cooperative Oncology Group (ECOG) Scoring können verwendet werden, um die Performance zu bewerten. Diese sind jedoch altersbedingt und daher nicht ausreichend, um Komorbidität und Gebrechlichkeit zu beurteilen. Der Charlson Komorbiditätsindex wurde von Sorror für Patienten, die sich einer allogenen, hämatopoietischen Stammzelltransplantation (allo-HSCT) unterziehen, angepasst (25, 26) und wurde auch für MDS-Patienten validiert (HCT-CI) (27). Gebrechlichkeit und Funktionalität im täglichen Leben kann mit allgemeinen geriatrischen Bewertungsinstrumenten bewertet werden. Basierend auf einer steigenden Krebsinzidenz bei älteren Patienten sowie einer steigenden Anzahl von zielgerichteten Therapien, ist die Beurteilung patientenbezogener Faktoren ein zunehmendes Erfordernis für eine geeignete Behandlungszuteilung.
MDS-Patienten mit niedrigem Risiko
Watch-and-wait
Die Lebenserwartung von Patienten > 70 Jahre mit MDS-SLD oder MDS mit del(5q) ist nicht signifikant kürzer, als diejenige einer altersangepassten, älteren Bevölkerung (24). Patienten mit niedrig/intermediär-1 IPSS und asymptomatischen Zytopenien sollten daher nur regelmässig kontrolliert werden ohne Behandlung (7). Diese Empfehlung könnte sich in Zukunft jedoch gegebenenfalls ändern, da mit NGS in dieser Patientengruppe auch ungünstige Mutationen nachgewiesen werden können, welche von einer früheren Behandlung profitieren.
Supportive Massnahmen
Die supportiven Massnahmen umfassen Transfusionen, Infektionsprophylaxe, Antiemetika, Analgetika, Eisenchelation und Wachstumsfaktoren. Eine Transfusionsabhängigkeit ist im Allgemeinen mit einem fortgeschrittenen Krankheitsstadium und einer schlechteren Prognose assoziiert (24). Erythrozytenkonzentrate (EKs) werden in der Regel ab einem Hämoglobinspiegel < 80 g / l (oder auf höherem Niveau, falls symptomatisch) transfundiert. Thrombozytenkonzentrate sollten nur zurückhaltend zur Prophylaxe von Blutungen eingesetzt werden. Üblicherweise steigt das Risiko spontaner Blutungen bei Thrombozyten < 5 - 10 G / L oder < 20 G / L mit zusätzlichen Risikofaktoren wie Fieber oder Mukositis. Transfusionen sind in der Regel mit einem höheren Risiko für unerwünschte Ereignisse wie Alloimmunisierung und transfusionsassoziierte Komplikationen assoziiert (28). EKs müssen bei Patienten, die potentielle Kandidaten für eine allo-HSCT sind, bestrahlt werden, um das Risiko einer HLA-Alloimmunisierung und transfusionsassoziierten Graft-versus-Host-Erkrankung zu reduzieren.
Bei stark transfundierten Patienten (> 20 - 25 EKs) und/oder Ferritinwerten > 1000 mg / l kann eine Chelation eine Eisenüberladung vorbeugen, Zytopenien in ca. einem Drittel der Patienten verbessern und möglicherweise auch das Gesamtüberleben günstig beeinflussen (29 - 31). Weiterhin bleibt aber die Frage nicht sicher geklärt, welche Patienten cheliert werden sollen, da bei MDS Patienten (noch) keine randomisierten Studien vorliegen (32). Eine Eisenchelation wird im Allgemeinen mit Deferasirox Patienten angeboten, welche Kandidaten für eine allo-HSCT sind oder eine Lebenserwartung von > 1 Jahr haben.
Patienten mit niedrig/intermediär-1 IPSS, mit Hämoglobinwerten < 100 g / l, Serum-Erythropoietin-Werten < 200 - 500 U/L, EK transfusionsunabhängig oder mit < 2 EKs/Monat sind Kandidaten für eine Behandlung mit Erythropoietin Stimulierenden Agenzien (ESA) (29, 33, 34). Zwischen den verschiedenen ESA-Produkten (rekombinantes humanes Erythropoietin (rHuEPO) Alpha und Beta oder Darbepoietin Alpha) wurden keine relevanten Unterschiede festgestellt. Die erforderlichen EPO-Dosierungen sind höher als bei Patienten mit Niereninsuffizienz und man beginnt in der Regel mit 30 000 U / Woche rHuEPO sc (ca. 150 μg Darbepoietin alpha). Bei fehlendem Ansprechen wird die Dosis nach 6-8 Wochen verdoppelt. Der zusätzliche Einsatz von Granulozyten-Colony Stimulaing Factor (G-CSF 3 x 300 - 480 ug / Woche sc) bei Patienten, die nicht genügend auf ESA ansprechen, ist kontrovers und wird in der Schweiz selten eingesetzt (29, 35, 36). Die Einhaltung eines strikten ESA/G-CSF-Substitutionsregimes ist wichtig, um frühzeitig refraktäre Patienten zu identifizieren, die eine schlechtere Prognose haben und allfällige Kandidaten für weiterführende Behandlungen sein könnten.
Thrombopoietin-stimulierende Agenzien (TSA) (Romiplostim, Eltrombopag) wurden in klinischen Studien bei Patienten mit Thrombozytopenie und niedrig/intermediär-1 IPSS getestet. TSA zeigen eine positive Wirkung auf die Thrombozytenzahl und können Blutungen reduzieren haben aber keinen Einfluss auf das Gesamtüberleben (37, 38). Es ist wichtig zu beachten, dass die Behandlungen mit Wachstumsfaktoren bei MDS-Patienten grundsätzlich zugelassen, aber «off-limitatio» sind und daher eine Kostengutsprache von der zuständigen Krankenkasse notwendig ist.
Die immunmodulatorische und anti-angiogenetische Wirkung von Thalidomid wurde schon früher bei MDS Patienten eingesetzt, um den Transfusionsbedarf zu reduzieren (39). Aufgrund der neurologischen Nebenwirkung von Thalidomid, wurde ein 4-Amino-glutarimid Derivat, das Lenalidomid (Revlimid®) ohne diesen ungünstigen Nebeneffekt entwickelt. Mit Lenalidomid lässt sich eine anhaltende Transfusionsunabhängigkeit und zytogenetische Remission bei etwa der Hälfte aller MDS-Patienten mit niedrig / intermediär-1 IPSS und isoliert del(5q) erreichen (40). In einer Phase-3-Studie fand man zudem auch in ca. einem Viertel der Nicht-del(5q)-MDS-Patienten mit niedrig/intermediär-1 IPSS eine Transfusionsunabhängigkeit, während Mutationen in TP53 mit Resistenz und einer Krankheitsprogression assoziiert waren (41, 42) und daher bei fehlendem Ansprechen Abklärungen in Richtung einer Transplantation rechtfertigen. 10% der MDS-Patienten präsentieren sich mit hypoplastischem Knochenmark und sind potentielle Kandidaten für eine immunsuppressive Behandlung mit Antithymozytenglobulin (ATG) in Kombination mit Cyclosporin A (CyA) mit Ansprechraten von etwa einem Drittel (43). Eine Kombination CyA/ATG mit dem TSA Eltrombopag hat einen zusätzlichen Nutzen bei Patienten mit aplastischer Anämie gezeigt, ist aber für hypoplastische MDS Patienten nicht zugelassen (44).
MDS-Patienten mit höherem Risiko
Hypomethylierende Agenzien
MDS-Patienten mit Blastenexzess oder mit höherem Risiko, welche für eine intensive Chemotherapie und allo-HSCT nicht in Frage kommen, sind Kandidaten für eine palliative Behandlung mit hypomethylierende Agenzien (HMAs). Die Pyrimidin-Nukleosid-Analoga, 5-Azacytidin (AZA) und 5 - Aza - 2’ - desoxycytidin / Decitabine (DEC), wurden in Phase-3-Studien an MDS Patienten mit höherem Risiko untersucht (45,46). HMAs sind im Allgemeinen gut verträglich und zeigten signifikant höhere partielle und vollständige Remissionen im Vergleich zu best supportive care, einschliesslich Hydroxyurea und niedrigdosiertem Cytosin Arabinosid (AraC). HMAs bleiben jedoch der intensivierten Induktions-Chemotherapie gefolgt von allo-HSCT unterlegen, für welche jedoch nur eine Minderheit der älteren MDS Patienten in Frage kommt. MDS mit komplexem Karyotyp, sollten aufgrund der niedrigeren Raten vollständiger Remissionen und der höheren Toxizität mit intensiven Chemotherapien, bevorzugt mit HMA behandelt werden (47). Es gibt keine allgemein anerkannten prädiktiven molekularen Marker für das Ansprechen auf HMA und auch die Dauer der Behandlung bleibt unklar. Derzeit gibt es keine etablierte Behandlung nach Versagen von HMAs, diese Patienten sollten daher vorzugsweise auf klinische Studien behandelt werden.
Intensive Induktionschemotherapie
Die AML-basierte Induktions-Chemotherapie, gefolgt von einer allo-HSCT ist MDS Patienten mit höherem Risiko vorbehalten, die für eine intensive Therapie genügend fit sind. Jüngeres Alter, guter Leistungsstatus und günstige Zytogenetik sind unabhängige prognostische Faktoren, die mit einem besseren Überleben assoziiert sind (48). Patienten mit ungünstigen oder komplexen zytogenetischen Veränderungen sowie Mutationen oder Deletionen in TP53 haben ein schlechteres Ansprechen auf eine intensive Chemotherapie und können von einer Behandlung mit HMA mit oder ohne anschliessender allo-HSCT profitieren (47, 49). Die derzeitige Datenlage ist aber im Allgemeinen noch nicht ausreichend, um HMA für die Induktion vor allo-HSCT ausserhalb klinischer Studien zu empfehlen (8).
Die allo HSCT bleibt die einzige kurative Option, ist aber nur für MDS-Patienten geeignet, die genügend fit sind. Die Beurteilung der Komorbiditäten ist wichtig für die Entscheidungsfindung, welche Patienten für eine allo-HSCT in Frage kommen und der HCT-CI Score wird oft für diesen Zweck verwendet (26). Für eine allo HSCT kommen MDS Patienten mit intermediär-2/hoch IPSS in Frage. Das Alter ist der wichtigste prädiktive Faktor für das Gesamtüberleben. Eine retrospektive Analyse der Europäischen Gruppe für Blut- und Knochenmarktransplantation (EBMT) ergab eine behandlungsassoziierte Mortalität von 30% bei Patienten < 20 Jahre, 43% bei Patienten zwischen 20 und 40 Jahren und 50% bei Patienten> 50 Jahre. Reduktion der Intensität der Konditionierung und sorgfältige Auswahl der Patienten haben jedoch gezeigt, dass eine allo-HSCT auch bei Patienten zwischen 60 und 70 Jahren möglich ist. Für MDS-Patienten, die aufgrund von Komorbiditäten nicht für eine myeloablative Konditionierung qualifizieren, kann eine Konditionierung mit reduzierter Intensität in Betracht gezogen werden, vorzugsweise innerhalb klinischer Studien.
Zukunftsperspektiven
Die Entdeckung von RSDM bei Patienten mit myeloischen Neoplasien eröffnet eine ungeahnte Palette neuer diagnostischer und therapeutischer Möglichkeiten. Eine klonale Evolution kann damit bereits in frühen Stadien einer klonalen Hämatopoese indentifiziert werden und erlaubt möglicherweise auch die Identifikation von Patienten, welche von einer früheren Intervention profitieren können. Es konnte zum Beispiel gezeigt werden, dass Mutationen in TP53, EZH2, ETV6, RUNX1 und ASXL1 mit einer schlechten Prognose assoziiert sind und ein molekulares Scoring-System wird derzeit entwickelt (IPSS-R Mole) (50). Bei MDS-Patienten mit niedrigem Risiko und RS- oder SF3B1-Mutationen (MDS-RS-SLD / MLS) führt der TGF-beta ligand-trap Luspatercept in zwei Drittel der Patienten zu einem erythroiden Ansprechen und Transfusionsfreiheit. Luspatercept scheint die Erythropoese durch Mechanismen zu verbessern, die unabhängig von EPO sind. Basierend auf der Annahme, dass eine niedrig dosierte und längere Exposition mit HMAs zu einer Verbesserung der Differenzierung führen kann, werden zur Zeit orale AZA-Formulierungen in klinischen Studien an MDS-Patienten mit niedrigem Risiko getestet (51). Weitere Medikamente, die an MDS Patienten untersucht werden, umfassen Toll-like-Rezeptor-2-Antikörper, CD95-Ligand(FAS-Ligand)-Hemmer, Multikinase-Inhibitoren (z. B. Rigosertib), Checkpoint-Inhibitoren (z. B. Nivolumab, Durvalumab), Telomerase-Inhibitoren (z. B. Imetelstat) und Inhibitoren von mutiertem IDH1 / 2 (zB AG - 120 / AG-221).
Darüber hinaus ist die Verbesserung der Behandlungsallokation basierend auf Wirksamkeit, Verträglichkeit, Nutzen und Richtlinien-Konformität ein aktives Forschungsgebiet der Versorgungsforschung. Daher ist der Einschluss von MDS-Patienten in longitudinale Kohortenstudien sehr zu begrüssen, so wie es die SAKK 33/18 («I-CARE for MDS») Studie verfolgen wird.
Weiter werden longitudinale Kohorten und Biobanking von biologischem Material von Patienten mit MDS oder frühen Formen einer klonalen Hämatopoese (CCUS) sehr nützlich sein, um detaillierte, gesundheitsbezogene Daten mit Biomarkern zu verknüpfen. Diese Plattform steht seit 2016 mit dem Swiss MDS Regsitry/Biobank zur Verfügung und soll in Zukunft helfen, unser Verständnis der MDS Biologie zu verbessern, um damit auch im Sinne einer «Präzisionsmedizin» die Prognose und das Therapieansprechen der Patienten besser abschätzen zu können.
Zusammenfassung
Die Myelodysplastischen Syndrome (MDS) bilden eine heterogene Gruppe von klonalen Erkrankungen des Blutes mit einer zunehmenden Inzidenz in der älteren Bevölkerung. Aufgrund der demographischen Alterung unserer Gesellschaft werden MDS eine wachsende Bedeutung in unserem Gesundheitswesen haben. MDS werden durch Genmutationen in den hämatopoetischen Stammzellen verursacht und sind gekennzeichnet durch eine ineffektive Hämatopoiese mit Zytopenien und Dysplasien sowie einer Neigung zur Progression in eine sekundäre akute myeloische Leukämie (sAML). Eine exakte Diagnose und Risikostratifizierung sind für eine korrekte Behandlung entscheidend. Patienten mit niedrigem Risiko haben ein medianes Überleben von 3 bis 8 Jahren und sterben vorwiegend an zytopenieassoziierten Komplikationen (kardiovaskuläre Ereignisse, Infektionen und Blutungen). Die Ziele einer Behandlung von Patienten mit niedrigem Risiko liegen daher vor allem in der Verbesserung der Lebensqualität, Verringerung von zytopenieassoziierten Komplikationen und einem Hinauszögern eines Progresses in ein höhergradiges MDS. Bei MDS Patienten mit hohem Risiko liegt hingegen die mediane Überlebenszeit nur bei 1 bis 3 Jahren und die AML-assoziierte Mortalität steht im Vordergrund. Bei diesen Patienten sollte die Behandlung darauf ausgerichtet sein, die Progression in eine AML hinauszuzögern und das Gesamtüberleben zu verbessern. Die allogene hämatopoetische Stammzelltransplantation bleibt die einzige kurative Option für Patienten mit hohem Risiko. Jedoch ist nur eine Minderheit der meistens älteren und polymorbiden MDS Patienten geeignet für eine solche intensive Behandlung. Daher werden die meisten Patienten mit supportiven Massnahmen und palliativen Behandlungen, wie zum Beispiel Wachstumsfaktoren, Immunmodulatoren und hypomethylierenden Agenzien behandelt. Da ältere Patienten mit chronischen Zytopenien häufig in der allgemein internistischen Praxis gesehen werden, ist die Kenntnis über mögliche Präsentationsformen und angemessene Behandlungsoptionen wichtig für alle Ärzte aus der Grundversorgung.
PD Dr. med. Nicolas Bonadies
Leitender Arzt und Koordinator Swiss MDS Study Group
Universitätsklinik für Hämatologie und Hämatologisches Zentrallabor
Inselspital Bern
Freiburgstrasse 18
3010 Bern
Nicolas.Bonadies@insel.ch
Der Autor hat deklariert, keine Interessenskonflikte in Zusammenhang mit diesem Artikel zu haben.
Die Myelodysplastischen Syndrome (MDS) bilden eine heterogene Gruppe von klonalen Erkrankungen des Blutes mit einer zunehmenden Inzidenz in der älteren Bevölkerung
MDS werden durch Genmutationen in den hämatopoetischen Stammzellen verursacht und sind gekennzeichnet durch eine ineffektive Hämatopoiese mit Zytopenien und Dysplasien sowie einer Neigung zur Progression in eine akute myeloische Leukämie (AML).
Eine exakte Diagnose und Risikostratifizierung sind für eine korrekte Behandlung entscheidend
Patienten mit niedrigem Risiko haben ein medianes Überleben von 3 bis 8 Jahren und sterben vorwiegend an zytopenieassoziierten Komplikationen (kardiovaskuläre Ereignisse, Infektionen und Blutungen).
Die Ziele einer Behandlung von Patienten mit niedrigem Risiko liegen daher vor allem in der Verbesserung der Lebensqualität, Verringerung von Zytopenie-assoziierten Komplikationen und einem Hinauszögern eines Progresses in ein höhergradiges MDS.
Bei MDS Patienten mit hohem Risiko liegt hingegen die mediane Überlebenszeit nur bei 1 bis 3 Jahren und die AML-assoziierte Mortalität steht im Vordergrund. Bei diesen Patienten sollte die Behandlung darauf ausgerichtet sein, die Progression in eine AML hinauszuzögern und das Gesamtüberleben zu verbessern.
Die allogene hämatopoetische Stammzelltransplantation bleibt die einzige kurative Option für Patienten mit hohem Risiko. Jedoch ist nur eine Minderheit der meistens älteren und polymorbiden MDS Patienten geeignet für eine solche intensive Behandlung
Take-Home Message
Les syndromes myélodysplasiques (MDS) sont un groupe hétérogène de maladies clonales du sang avec une incidence croissante chez les personnes âgées.
Les MDS sont causés par des mutations génétiques dans les cellules souches hématopoïétiques et se caractérisent par une hématopoïèse inefficace avec une cytopénie et une dysplasie et une tendance à la progression vers la leucémie myéloïde aiguë (LMA).
Le diagnostic exact et la stratification des risques sont cruciaux pour un traitement correct.
Les patients à faible risque ont une survie médiane de 3 à 8 ans et meurent principalement de complications associées à la cytopénie (événements cardiovasculaires, infections et saignements).
Les principaux objectifs du traitement des patients à faible risque sont donc d’améliorer la qualité de vie, de réduire les complications associées à la cytopénie et de retarder l’évolution vers un MDS de grade supérieur.
Chez les patients atteints de MDS à risque élevé, cependant, le temps de survie médian n’est que de 1 à 3 ans et la mortalité associée à la LMA est au premier plan. Chez ces patients, le traitement doit être conçu de manière à retarder la progression vers la LMA et à améliorer la survie globale.
La greffe de cellules souches hématopoïétiques allogéniques demeure la seule option curative pour les patients à haut risque. Cependant, seule une minorité des patients atteints de MDS généralement âgés et polymorbides convient à ce type de traitement intensif.
1. Bonadies N, Feller A, Rovo A, et al. Trends of classification, incidence, mortality, and survival of MDS patients in Switzerland between 2001 and 2012. Cancer Epidemiol 2017;46:85-92.
2. Dinmohamed AG, Visser O, van Norden Y, et al. Trends in incidence, initial treatment and survival of myelodysplastic syndromes: a population-based study of 5144 patients diagnosed in the Netherlands from 2001 to 2010. Eur J Cancer 2014;50:1004-12.
3. Roman E, Smith A, Appleton S, et al. Myeloid malignancies in the real-world: Occurrence, progression and survival in the UK’s population-based Haematological Malignancy Research Network 2004-15. Cancer Epidemiol 2016;42:186-98.
4. Neukirchen J, Schoonen WM, Strupp C, et al. Incidence and prevalence of myelodysplastic syndromes: data from the Dusseldorf MDS-registry. Leuk Res 2011;35:1591-6.
5. Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer 2007;7:118-29.
6. Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:504-15.
7. Malcovati L, Hellstrom-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 2013;122:2943-64.
8. Abdel-Wahab O, Figueroa ME. Interpreting new molecular genetics in myelodysplastic syndromes. Hematology American Society of Hematology Education Program 2012;2012:56-64.
9. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011;364:2496-506.
10. Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011;478:64-9.
11. Kon A, Shih LY, Minamino M, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 2013;45:1232-7.
12. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391-405.
13. Valent P, Horny HP. Minimal diagnostic criteria for myelodysplastic syndromes and separation from ICUS and IDUS: update and open questions. Eur J Clin Invest 2009;39:548-53.
14. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477-87.
15. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488-98.
16. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9-16.
17. Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001-2004, using data from the NAACCR and SEER programs. Blood 2008;112:45-52.
18. Santini V. Treatment of low-risk myelodysplastic syndromes. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program 2016;2016:462-9.
19. Fenaux P, Ades L. How we treat lower-risk myelodysplastic syndromes. Blood 2013;121:4280-6.
20. Komrokji RS. Current State of the Art: Management of Higher Risk Myelodysplastic Syndromes. Clinical lymphoma, myeloma & leukemia 2016;16 Suppl:S39-43.
21. Garcia-Manero G. Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management. American journal of hematology 2015;90:831-41.
22. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997;89:2079-88.
23. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012;120:2454-65.
24. Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2007;25:3503-10.
25. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis 1987;40:373-83.
26. Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 2005;106:2912-9.
27. Sorror ML, Storb RF, Sandmaier BM, et al. Comorbidity-age index: a clinical measure of biologic age before allogeneic hematopoietic cell transplantation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2014;32:3249-56.
28. Szczepiorkowski ZM, Dunbar NM. Transfusion guidelines: when to transfuse. Hematology American Society of Hematology Education Program 2013;2013:638-44.
29. Malcovati L, Hellstrom-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 2013;122:2943-64.
30. Gattermann N, Finelli C, Della Porta M, et al. Hematologic responses to deferasirox therapy in transfusion-dependent patients with myelodysplastic syndromes. Haematologica 2012;97:1364-71.
31. List AF, Baer MR, Steensma DP, et al. Deferasirox reduces serum ferritin and labile plasma iron in RBC transfusion-dependent patients with myelodysplastic syndrome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30:2134-9.
32. Mitchell M, Gore SD, Zeidan AM. Iron chelation therapy in myelodysplastic syndromes: where do we stand? Expert Rev Hematol 2013;6:397-410.
33. Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood 2008;111:574-82.
34. Hellstrom-Lindberg E, Negrin R, Stein R, et al. Erythroid response to treatment with G-CSF plus erythropoietin for the anaemia of patients with myelodysplastic syndromes: proposal for a predictive model. British journal of haematology 1997;99:344-51.
35. Balleari E, Rossi E, Clavio M, et al. Erythropoietin plus granulocyte colony-stimulating factor is better than erythropoietin alone to treat anemia in low-risk myelodysplastic syndromes: results from a randomized single-centre study. Ann Hematol 2006;85:174-80.
36. Casadevall N, Durieux P, Dubois S, et al. Health, economic, and quality-of-life effects of erythropoietin and granulocyte colony-stimulating factor for the treatment of myelodysplastic syndromes: a randomized, controlled trial. Blood 2004;104:321-7.
37. Kantarjian H, Fenaux P, Sekeres MA, et al. Safety and efficacy of romiplostim in patients with lower-risk myelodysplastic syndrome and thrombocytopenia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2010;28:437-44.
38. Mavroudi I, Pyrovolaki K, Pavlaki K, et al. Effect of the nonpeptide thrombopoietin receptor agonist eltrombopag on megakaryopoiesis of patients with lower risk myelodysplastic syndrome. Leuk Res 2011;35:323-8.
39. Raza A, Meyer P, Dutt D, et al. Thalidomide produces transfusion independence in long-standing refractory anemias of patients with myelodysplastic syndromes. Blood 2001;98:958-65.
40. Fenaux P, Giagounidis A, Selleslag D, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011;118:3765-76.
41. Santini V, Almeida A, Giagounidis A, et al. Randomized Phase III Study of Lenalidomide Versus Placebo in RBC Transfusion-Dependent Patients With Lower-Risk Non-del(5q) Myelodysplastic Syndromes and Ineligible for or Refractory to Erythropoiesis-Stimulating Agents. J Clin Oncol 2016;34:2988-96.
42. Jadersten M, Saft L, Smith A, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:1971-9.
43. Passweg JR, Giagounidis AA, Simcock M, et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: a prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care–SAKK 33/99. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:303-9.
44. Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med 2012;367:11-9.
45. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009;10:223-32.
46. Lubbert M, Suciu S, Baila L, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:1987-96.
47. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291-9.
48. Ossenkoppele GJ, Graveland WJ, Sonneveld P, et al. The value of fludarabine in addition to ARA-C and G-CSF in the treatment of patients with high-risk myelodysplastic syndromes and AML in elderly patients. Blood 2004;103:2908-13.
49. Knipp S, Hildebrand B, Kundgen A, et al. Intensive chemotherapy is not recommended for patients aged >60 years who have myelodysplastic syndromes or acute myeloid leukemia with high-risk karyotypes. Cancer 2007;110:345-52.
50. Nazha A, Narkhede M, Radivoyevitch T, et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 2016.
51. Saunthararajah Y. Key clinical observations after 5-azacytidine and decitabine treatment of myelodysplastic syndromes suggest practical solutions for better outcomes. Hematology American Society of Hematology Education Program 2013;2013:511-21.