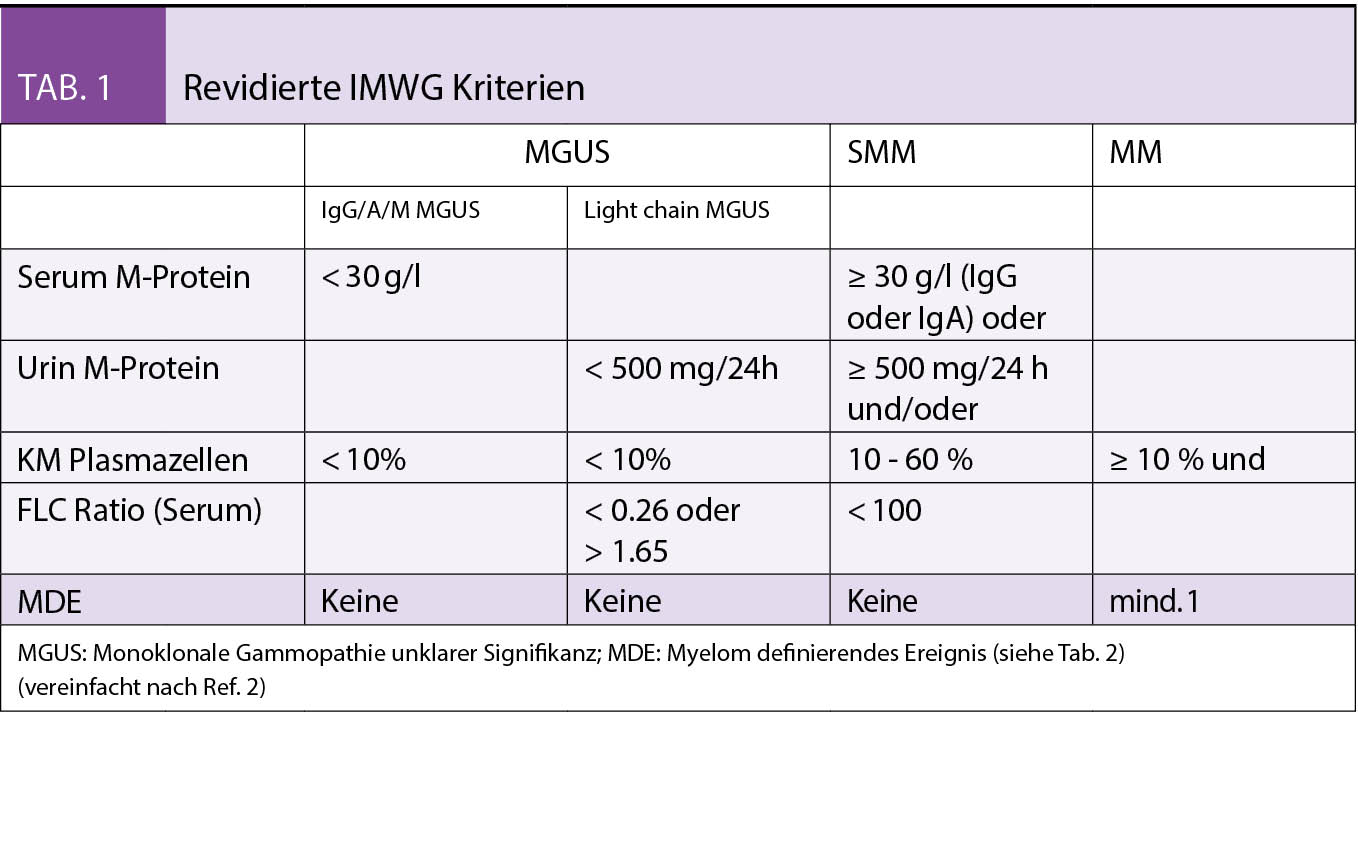
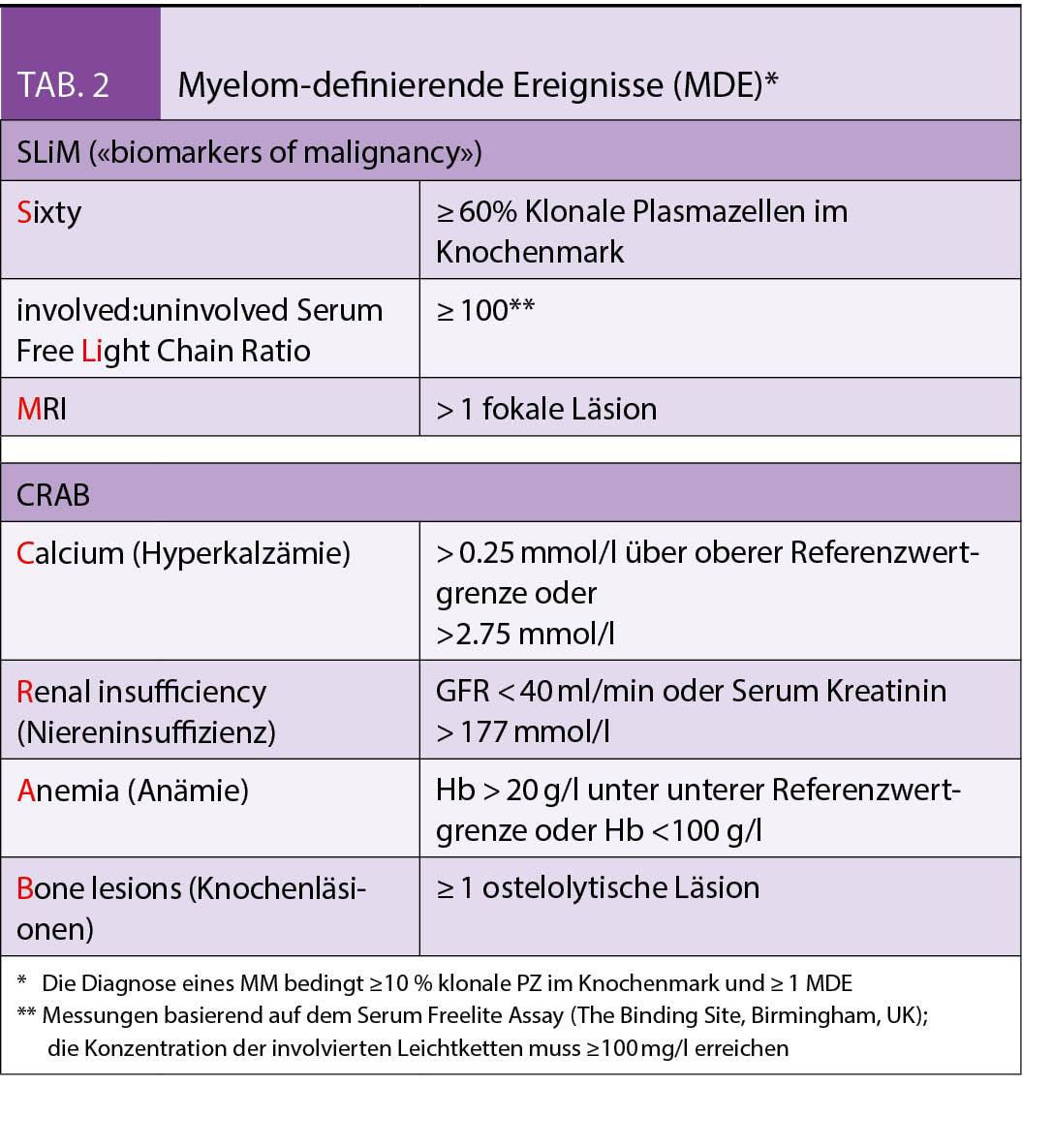
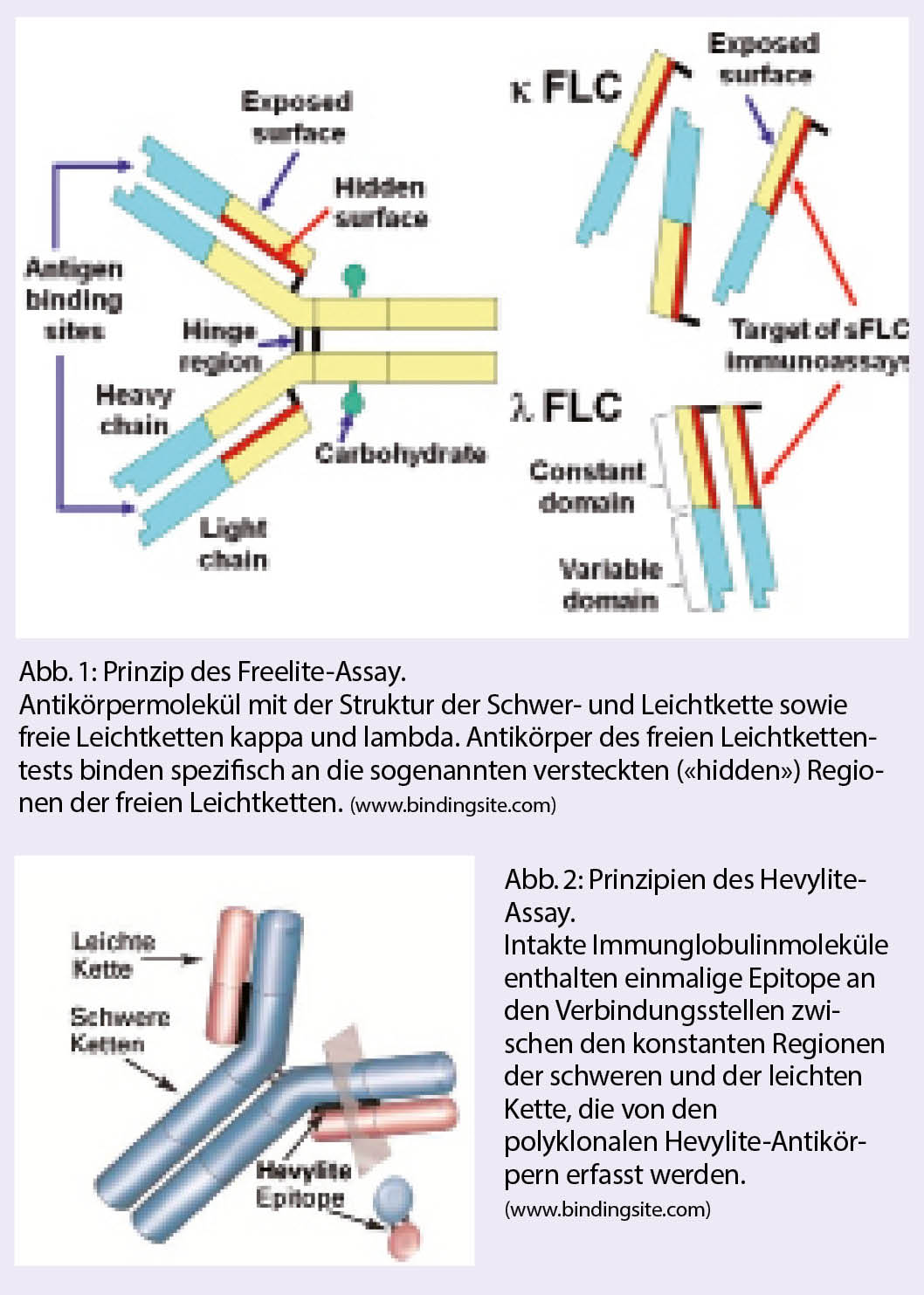
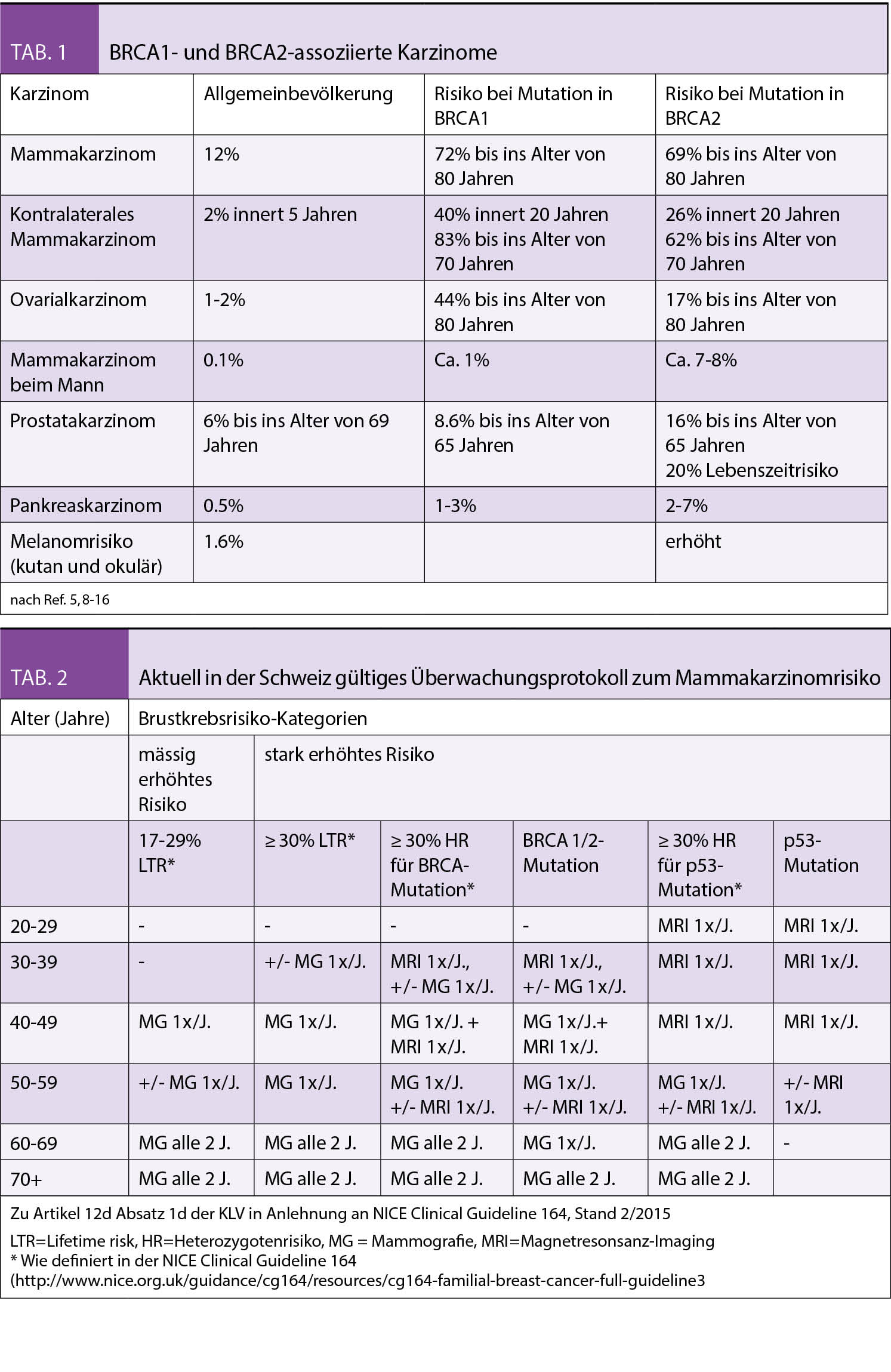
Die Personalisierte Onkologie hat dank der Entwicklung von modernsten diagnostischen Werkzeugen und dem dadurch ermöglichten, therapeutischen Einsatz zielgerichteter Therapien über die letzten Jahre stark an Bedeutung gewonnen. Gerade das nicht-squamöse, nicht-kleinzellige Bronchuskarzinom ist ein Paradebeispiel dafür.
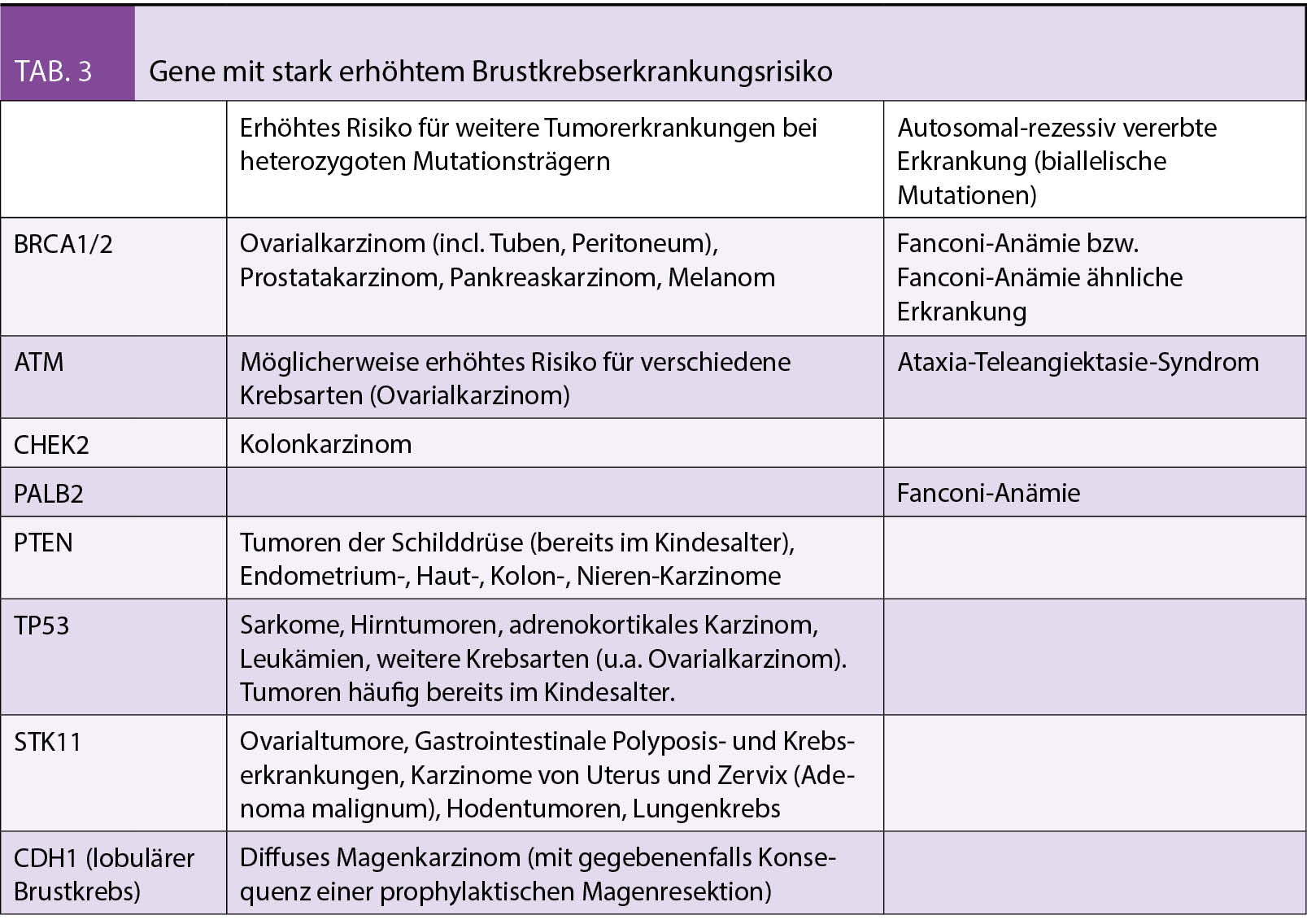
Zielgerichtete Therapien mit Tyrosinkinase-Inhibitoren (TKIs) haben im Zeitalter der Personalisierten Onkologie (oder «Precision Oncology») über die letzten Jahre eine zunehmende Bedeutung erhalten. Nach ersten Erfolgen in der chronisch myeloischen Leukämie (CML) mit dem Einsatz von Imatinib (1) und im BRAF p.V600E-mutierten Melanom mit BRAF- und MEK-TKIs (2–4) hat sich das nicht-squamöse, nicht-kleinzellige Lungenkarzinom (nsqNSCLC) als eigentliches Aushängeschild für den Einsatz zielgerichteter Therapien mit TKIs heraus kristallisiert. Das hängt einerseits mit der Vielfalt an entdeckten onkogenen Veränderungen zusammen, die einer zielgerichteten Therapie zugänglich sind (sogenannte «actionable aberrations»). Zusammen gezählt weisen rund 40% aller nsqNSCLC eine Aberration auf, für die eine in der Schweiz erhältliche zielgerichtete Therapie-Form existiert (Abb. 1) (5). Bei absolut rund 4’200 neu diagnostizierten Lungenkrebs-Fällen pro Jahr (rund 50% davon nsqNSCLC) betrifft das in der Schweiz theoretisch rund 800–900 Patienten (6). Andererseits hat die Weiterentwicklung der molekular-pathologischen Diagnostik mit Einführung der Tiefensequenzierung (NGS) im Rahmen von Panel-Testungen in der Routine dazu geführt, dass diese Aberrationen rasch und zuverlässig bestimmt werden können. Im Folgenden wollen wir die aktuell wichtigsten klinischen Daten zum Einsatz von TKIs im nsqNSCLC diskutieren.
EGFR-mutiertes NSCLC
Aktivierende Mutationen im EGFR-Gen stellen die häufigste onkogene genomische Aberration dar, für die eine zielgerichtete Therapie existiert. In über 90% der Fälle handelt es sich um eine sogenannte klassische EGFR-Mutation (EGFR p.L858R oder eine Deletion im Exon 19). Mehrere randomisierte Studien haben in dieser Situation die Überlegenheit einer zielgerichteten Therapie gegenüber einer Chemotherapie gezeigt und drei EGFR-TKIs der ersten und zweiten Generation sind seither Standard in first-line: Erlotinib (Erlo), Gefitinib (Gefi) und Afatinib (Afa) (7–12). Bei den nicht klassischen («uncommon») EGFR-Mutationen wird bevorzugt Afa eingesetzt, da es dort am besten dokumentierte Aktivität hat (13).
Über kurz oder lang werden die meisten Patienten eine Resistenz auf die EGFR-TKI-Behandlung entwickeln. Der häufigste Mechanismus ist das Auftreten einer EGFR p.T790M-Punktmutation, weitere Beispiele sind MET-Amplifikationen, HER2-Amplifikationen, epitheliale-zu-mesenchymale Transition, oder sogar Entwicklung einer kleinzelligen Histologie. EGFR-TKIs der dritten Generation sind gegen EGFR p.T790M wirksam, allen voran Osimertinib (Osi). Die randomisierte Phase-3 Studie AURA3 hat einen deutlichen PFS-Vorteil für Osi gegenüber einer Platin/Pemetrexed (Pem)-Kombination gezeigt (10.1 vs 4.4 Mt; HR 0.30 [95% CI 0.23–0.41]; P < 0.001) (14). Beim Auftreten von klinischer Resistenz gegenüber einer EGFR-TKI-Therapie soll daher immer aktiv nach EGFR p.T790M gesucht werden, sei es in einer liquid biopsy (Analyse an zirkulierender, freier Tumor-DNS) oder an einer Biopsie eines progredienten Herdes. Wir verfolgen im Allgemeinen die Strategie beides gleichzeitig zu testen, da die liquid biopsy nicht konklusiv sein kann (nämlich dann, wenn der Tumor zu wenig DNS in die Zirkulation abgibt), und da wegen intra- und intertumoraler Heterogenität eine Biopsie falsch negativ sein kann. Durch die doppelte Analyse kann man somit das Risiko minimieren, eine T790M zu verpassen.
Osi wurde in der Zwischenzeit in der Phase-3 Studie FLAURA auch in first-line untersucht, randomisiert gegen Erlo oder Gefi (15). Osi hat bei noch nicht reifen OS-Daten zu einer deutlichen PFS-Verlängerung geführt, so dass es inzwischen hierfür die Zulassung erhalten hat und zum neuen Standard werden dürfte (18.9 vs 10.2 Mt; HR 0.46 [95% CI 0.37–0.57]; P < 0.001). Vor allem im ZNS ist Osi aktiver als die TKIs der ersten Generation (16).
Es gibt verschiedene weitere Ansätze, wie die Therapie in first-line verbessert werden könnte. Einer ist die Hinzugabe des VEGF-gerichteten, monoklonalen Antikörpers Bevacizumab (Beva), infolge präklinischer Hinweise auf einen möglichen Synergismus (17). Die randomisierte japanische JO25567 Studie und die einarmige Phase-2 Studie ETOP 2-11 BELIEF haben beide eine eindrückliche Verlängerung des PFS auf rund 16 Mt gezeigt (18, 19). Der Enthusiasmus wurde allerdings am diesjährigen Jahreskongress der Amerikanischen Krebsgesellschaft ASCO gedämpft: etwas überraschend hat die PFS-Verlängerung in der finalen Analyse nicht zu einer OS-Verlängerung geführt (20).
Ein anderer interessanter Ansatz ist die Kombination eines EGFR-TKIs mit Chemotherapie. In der Vergangenheit waren solche Studien negativ geblieben, hatten diese Kombination allerdings in anderen Situationen untersucht: Im Rahmen der INTACT-2 Studie in einer unselektionierten NSCLC-Population (21) und im Rahmen der IMPRESS-Studie in second-line nach Gefi Versagen (22). Ebenfalls am ASCO meeting 2018 wurden jetzt aber erste Resultate der NEJ009-Studie präsentiert, welche randomisiert Gefi mono vs Gefi / Carboplatin (Carbo) / Pem in first-line bei EGFR-mutierten Patienten untersuchte. Die Hinzugabe der Chemotherapie hat das OS statistisch signifikant um mehr als ein Jahr verlängert (52.2 vs 38.8 Mt; (HR 0.70 [95% CI 0.52–0.93]; p = 0.013) (23). Es wäre nun spannend zu untersuchen, welche Wirksamkeit eine Kombination von Chemotherapie mit dem derzeit aktivsten EGFR-TKI Osi in first-line hat.
ALK-transloziertes NSCLC
Anders als beim EGFR-mutierten NSCLC ist der Mechanismus der onkogenen Aktivierung bei ALK getriebenen NSCLC: hier handelt es sich um Translokationen mit Bildung eines Fusions-Onkoproteins, in den allermeisten Fällen EML4-ALK als Folge einer Inversion inv(2)(p21p23) (24). Dies führt zu einer konstitutiven Aktivierung der ALK-Kinase mit Hyperaktivierung von nachgeschalteten intrazellulären Signalwegen (25). ALK-Inhibitoren können dies therapeutisch ausnützen und der Archetyp ist Crizotinib (Crizo). Im Vergleich zu Standard-Chemotherapie hat Crizo eine deutliche PFS-Verlängerung gezeigt, zuerst in second-line (PROFILE 1007 Studie), dann auch in first-line (PROFILE 1014; medianes PFS 10.9 vs 7.0 Mt; HR 0.45 [95% CI 0.35–0.60]; P < 0.001) (26, 27). Die abschliessende OS-Analyse wurde kürzlich publiziert und zeigt rund 46 Mt in beiden Armen (ns), allerdings haben 84.2% der Patienten aus dem Chemotherapie-Arm in späteren Therapielinien Crizo erhalten (28). Wird für diesen crossover korrigiert, zeigt sich ein deutlicher OS-Vorteil für Crizo (HR 0.35; 95% bootstrap CI 0.08–0.72). Überdies war das OS in derjenigen Subgruppe am längsten, die Crizo und weitere ALK TKIs erhalten hat. Diese Daten sprechen klar dafür, dass im ALK-translozierten NSCLC zunächst eine ALK-Tyrosinkinase-Inhibition eingesetzt werden soll.
Auch im ALK-translozierten NSCLC kommt es allerdings zu Resistenzentwicklungen. Ein häufiger Mechanismus sind Punktmutationen in ALK, wobei es im Unterschied zu EGFR keine einzelne Mutation gibt, die klar überwiegt. Mehrere TKIs der nächsten Generation haben gezeigt, dass sie eine Crizo-Resistenz überwinden können: Alectinib (Alec), Ceritinib (Ceri), Lorlatinib (Lorla) und Brigatinib (Briga), wovon die ersten beiden in der Schweiz zugelassen sind (29–33). Auch beim ALK-translozierten NSCLC wurde dann als nächstes untersucht, ob die effizienteren ALK-Inhibitoren in first-line eingesetzt das outcome weiter verbessern können. Eine Überlegung dahinter ist, dass so die Entwicklung von Hirnmetastasen aufgeschoben oder gar verhindert werden kann. Hirnmetastasen sind bei ALK-translozierten NSCLC häufig und Crizo hat eine nur ungenügende Aktivität im ZNS. Ein weiteres Argument für einen Einsatz des effizientesten Medikamentes in first-line ist die Tatsache, dass man von Therapie-Linie zu Therapie-Linie immer Patienten verliert und somit nicht alle die Chance auf den effizientesten TKI erhalten, wenn man ihn sich für spätere Linien «aufspart». So hat Ceri gezeigt, dass es in first-line effizient ist – allerdings im Vergleich mit Chemotherapie und nicht mit dem Goldstandard Crizo (34). Letzten Sommer wurden dann auch die Resultate der ALEX-Studie voll publiziert, welche Alec gegen Crizo in unbehandelten Patienten untersucht hat (35). Das mediane PFS für Alec war noch nicht erreicht, aber die 12-Monats-Event-freie Rate war mit Alec signifikant höher (68.4% [95% CI, 61.0–75.9%] vs 48.7% [95% CI, 40.4–56.9%]; HR 0.47 [95% CI, 0.34–0.65]; P < 0.001). Ein Hauptgrund dafür war wie erwartet die wesentlich bessere ZNS-Kontrolle durch Alec mit einer kumulativen 12-Monats-Inzidenz einer ZNS-Progression von nur 9.4% (95% CI 5.4–14.7%) gegenüber 41.4% unter Crizo (95% CI 33.2–49.4%). Diese überzeugenden Daten haben auch zur Zulassung von Alec in first-line in der Schweiz geführt. Vor kurzem wurden auch erste Daten der ALTA-1L-Studie publiziert, welche ähnliche Resultate für Briga versus Crizo in der first-line zeigen konnte (36).
In der Praxis stellt sich nun natürlich die Frage nach der optimalen Behandlungssequenz bei dieser grossen Anzahl zugelassener TKIs. Im allgemeinen wird man je nach Zulassungsstatus mit einem möglichst effizienten ALK-TKI beginnen (Stand heute in der Schweiz wäre das Alec). Beim Auftreten von klinischer Resistenz würden wir dann empfehlen, eine Re-Biopsie anzustreben. Es ist bekannt, dass gewisse Resistenzmutationen in vitro unterschiedlich sensitiv gegenüber anderen TKIs sind, und dieses Sensitivitäts-Muster könnte die Auswahl des nächsten TKIs leiten (37). Prospektive, klinische Daten zum Vorteil dieses Ansatzes fehlen allerdings.
Andere Aberrationen
Es sind eine Reihe weiterer Aberrationen bekannt, für die es ebenfalls zielgerichtete Therapie-Optionen gibt. So zum Beispiel die BRAF p.V600E-Mutation – wie eingangs erwähnt bestens bekannt im Melanom. Gleich wie beim Melanom wurde auch beim BRAF-mutierten NSCLC eine Kombination aus BRAF- und MEK-Inhibitor untersucht, da dies in den Melanom-Studien die Resistenzentwicklung verzögern konnte und überdies besser verträglich war als eine reine BRAF-Inhibition (2-4). Eine doppelte TKI-Therapie (Dabrafenib/Trametinib) ist auch im nsqNSCLC sehr aktiv und hat zur Zulassung geführt. Sie stellt heute die Standardtherapie in second-line dar, in der EU auch in first-line (38).
Translokationen des ROS1-Gens sind eine weitere beschriebene Aberration. Interessanterweise sind eine Reihe unterschiedlicher Fusionspartner beschrieben worden, aber der onkogene Mechanismus ist eine Hyperaktivierung der ROS1-Kinase durch die Translokation und der Fusionspartner scheint dies nicht zu beeinflussen (39). Ebenso scheint die Sensitivität gegenüber ROS1-TKIs (welche alle auch Aktivität gegenüber ALK aufweisen) nicht vom Translokationspartner abzuhängen (39). Einzig Patienten mit der CD74-ROS1-Fusion scheinen häufiger Hirnmetastasen zu entwickeln. Da der erste bekannte ROS1-TKI Crizo war, hatte diese Gruppe ein tendenziell schlechteres Outcome (40). Inzwischen wurde aber gezeigt, dass bei ROS1-Translokationen auch Ceri und Lorla wirksam sind, welche bessere ZNS-Aktivität aufweisen (32,41).
Ebenfalls durch eine Translokation zum Onkogen wird das Proto-Onkogen RET – eine weitere Tyrosinkinase. Allerdings haben die bekannten TKIs eher bescheidene Aktivität, wie eine retrospektive Registeranalyse zeigen konnte (42). Prospektiv untersucht werden nun zur Zeit Alec (in der ETOP 12–17 ALERT Studie), sowie das Molekül LOXO-292 (LIBRETTO-001 Studie).
Anders ist der Mechanismus bei Veränderungen im cMET Gen. Hier treten eine Reihe von Mutationen auf, die wegen ihrer Vielfalt erst durch NGS-Techniken entdeckt werden konnten. Alle Mutationen haben aber gemeinsam, dass das Exon 14 des cMET Gens durch die splicing Maschinerie während der Transkription entfernt wird. Das führt dazu, dass im fertig translatierten cMET-Protein eine Domäne mit einer regulatorischen Phospho-Site fehlt (MET p.Y1003), welche ein Signal für eine Ubiquitin-Ligase darstellt. Ohne dieses Signal kann das Protein nicht effizient genug abgebaut werden und es kommt zu überaktiviertem MET-Signalling. Dies kann isoliert, wie auch in Kombination mit einer Amplifikation des cMET-Gens auftreten. Solche MET-Veränderungen wirken sensibilisierend gegenüber TKIs, wie zum Beispiel Crizo (43). Weitere Moleküle befinden sich zur Zeit in klinischen Studien (Tepotinib, Glesatinib oder Capmatinib).
Systemtherapie nach Ausschöpfen der zielgerichteten Therapie-Möglichkeiten
Zu guter Letzt stellt sich die Frage, wie man diejenigen Patienten am besten behandeln soll, bei denen die Möglichkeiten der TKI-Therapie(n) ausgeschöpft sind. Standard-Therapie ist hier sicher eine platin-basierte Kombinationschemotherapie, mit zum Beispiel Pem, falls diese nicht bereits als first-line Therapie vor einer TKI-Behandlung stattgefunden hat (44). Für Patienten mit EGFR-Mutationen konnte in Studien in second- und späteren Linien gezeigt werden, dass eine reine Immun-Checkpoint-Inhibition keinen Vorteil bringt, ganz im Gegensatz zu wild-typ Patienten (45–47). Es stellt sich allerdings die Frage, ob eine Chemo-/Immun-Therapie-Kombination einen Vorteil bringen könnte. In der dreiarmigen IMpower150 Studie waren Patienten mit EGFR- und ALK-Aberrationen explizit zugelassen (nach einer gehabten TKI-Linie). Die Studie hat die Hinzugabe von Atezolizumab (A) zum Chemotherapie-Backbone Carbo (C) / Paclitaxel (P) +/– Beva (B) in der first-line untersucht und der Vergleich ABCP versus BCP wurde dieses Jahr voll publiziert (48). Die Vierfachkombination hat dabei eine statistisch signifikante und klinisch bedeutsame Verlängerung des medianen PFS und OS gezeigt (PFS 8.3 vs. 6.8 Mt; HR 0.62; 95% CI 0.52–0.74; P < 0.001. OS 19.2 vs. 14.7 Mt; HR 0.78; 95% CI 0.64–0.96; P = 0.02). Dies nota bene auch in der EGFR- und ALK-positiven Subgruppe. Diese Therapie stellt somit sicherlich eine Alternative zur reinen Chemotherapie dar.
Schlussfolgerungen
Für Patienten mit onkogen aktivierenden Treibermutationen, welche therapeutisch angegangen werden können, gibt es sehr gute Therapie-Optionen, welche das Überleben um mehrere Monate verlängern können (49). Entscheidend ist allerdings, dass diese auch aktiv gesucht und zuverlässig diagnostiziert werden. Das bedingt eine stete, enge Diskussion zwischen medizinischen Onkologen und (Molekular)-Pathologen, idealerweise im Rahmen eines institutionalisierten, molekularen Tumorboards. Oftmals liegt nur wenig Gewebe für die Diagnostik vor und es ist daher essentiell, dass dieses auch ökonomisch eingesetzt wird. Hier können NGS-basierte Panel-Testungen einen wichtigen Beitrag leisten.
UniversitätsSpital Zürich
Klinik für Medizinische Onkologie und Hämatologie
Comprehensive Cancer Center Zurich
Rämistrasse 100
8091 Zürich
christian.britschgi@usz.ch
Klinikdirektorin
Klinik für Medizinische Onkologie
Freiburger Spital, HFR Kantonsspital Freiburg
1708 Fribourg
UniversitätsSpital Zürich
Klinik für Medizinische Onkologie und Hämatologie
Comprehensive Cancer Center Zurich
Rämistrasse 100
8091 Zürich
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Beitrag deklariert.
1. Hochhaus, A. et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. The New England journal of medicine 376, 917-927, doi:10.1056/NEJMoa1609324 (2017).
2. Dummer, R. et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. The Lancet. Oncology 19, 603-615, doi:10.1016/s1470-2045(18)30142-6 (2018).
3. Long, G. V. et al. Long-Term Outcomes in Patients With BRAF V600-Mutant Metastatic Melanoma Who Received Dabrafenib Combined With Trametinib. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, Jco2017741025, doi:10.1200/jco.2017.74.1025 (2017).
4. Larkin, J. et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. The New England journal of medicine 371, 1867-1876, doi:10.1056/NEJMoa1408868 (2014).
5. Tsao, A. S. et al. Scientific Advances in Lung Cancer 2015. J Thorac Oncol 11, 613-638, doi:10.1016/j.jtho.2016.03.012 (2016).
6. KrebsligaSchweiz. Krebs in der Schweiz: wichtige Zahlen. https://www.krebsliga.ch/ueber-krebs/zahlen-fakten/-dl-/fileadmin/downloads/sheets/zahlen-krebs-in-der-schweiz.pdf (2017).
7. Mok, T. S. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. The New England journal of medicine 361, 947-957, doi:10.1056/NEJMoa0810699 (2009).
8. Mitsudomi, T. et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. The Lancet. Oncology 11, 121-128, doi:10.1016/s1470-2045(09)70364-x (2010).
9. Maemondo, M. et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. The New England journal of medicine 362, 2380-2388, doi:10.1056/NEJMoa0909530 (2010).
10. Zhou, C. et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. The Lancet. Oncology 12, 735-742, doi:10.1016/s1470-2045(11)70184-x (2011).
11. Rosell, R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. The Lancet. Oncology 13, 239-246, doi:10.1016/s1470-2045(11)70393-x (2012).
12. Yang, J. C. et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. The Lancet. Oncology 16, 141-151, doi:10.1016/s1470-2045(14)71173-8 (2015).
13. Yang, J. C. et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. The Lancet. Oncology 16, 830-838, doi:10.1016/s1470-2045(15)00026-1 (2015).
14. Mok, T. S. et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. The New England journal of medicine 376, 629-640, doi:10.1056/NEJMoa1612674 (2017).
15. Soria, J. C. et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. The New England journal of medicine, doi:10.1056/NEJMoa1713137 (2017).
16. Reungwetwattana, T. et al. CNS Response to Osimertinib Versus Standard Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients With Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, Jco2018783118, doi:10.1200/jco.2018.78.3118 (2018).
17. Naumov, G. N. et al. Combined vascular endothelial growth factor receptor and epidermal growth factor receptor (EGFR) blockade inhibits tumor growth in xenograft models of EGFR inhibitor resistance. Clin Cancer Res 15, 3484-3494, doi:10.1158/1078-0432.ccr-08-2904 (2009).
18. Seto, T. et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomised, multicentre, phase 2 study. The Lancet. Oncology 15, 1236-1244, doi:10.1016/s1470-2045(14)70381-x (2014).
19. Rosell, R. et al. Erlotinib and bevacizumab in patients with advanced non-small-cell lung cancer and activating EGFR mutations (BELIEF): an international, multicentre, single-arm, phase 2 trial. The Lancet. Respiratory medicine 5, 435-444, doi:10.1016/s2213-2600(17)30129-7 (2017).
20. Yamamoto, N. et al. Erlotinib plus bevacizumab (EB) versus erlotinib alone (E) as first-line treatment for advanced EGFR mutation–positive non-squamous non–small-cell lung cancer (NSCLC): Survival follow-up results of JO25567. Journal of Clinical Oncology 36, 9007-9007, doi:10.1200/JCO.2018.36.15_suppl.9007 (2018).
21. Herbst, R. S. et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial–INTACT 2. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 22, 785-794, doi:10.1200/jco.2004.07.215 (2004).
22. Mok, T. S. K. et al. Gefitinib Plus Chemotherapy Versus Chemotherapy in Epidermal Growth Factor Receptor Mutation-Positive Non-Small-Cell Lung Cancer Resistant to First-Line Gefitinib (IMPRESS): Overall Survival and Biomarker Analyses. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 35, 4027-4034, doi:10.1200/jco.2017.73.9250 (2017).
23. Nakamura, A. et al. Phase III study comparing gefitinib monotherapy (G) to combination therapy with gefitinib, carboplatin, and pemetrexed (GCP) for untreated patients (pts) with advanced non-small cell lung cancer (NSCLC) with EGFR mutations (NEJ009). Journal of Clinical Oncology 36, 9005-9005, doi:10.1200/JCO.2018.36.15_suppl.9005 (2018).
24. Soda, M. et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561-566, doi:10.1038/nature05945 (2007).
25. Shaw, A. T. & Solomon, B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res 17, 2081-2086, doi:10.1158/1078-0432.ccr-10-1591 (2011).
26. Solomon, B. J. et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. The New England journal of medicine 371, 2167-2177, doi:10.1056/NEJMoa1408440 (2014).
27. Shaw, A. T. et al. Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer. New England Journal of Medicine 368, 2385-2394, doi:doi:10.1056/NEJMoa1214886 (2013).
28. Solomon, B. J. et al. Final Overall Survival Analysis From a Study Comparing First-Line Crizotinib Versus Chemotherapy in ALK-Mutation-Positive Non-Small-Cell Lung Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 36, 2251-2258, doi:10.1200/jco.2017.77.4794 (2018).
29. Shaw, A. T. et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. The Lancet. Oncology 17, 234-242, doi:10.1016/s1470-2045(15)00488-x (2016).
30. Ou, S. H. et al. Alectinib in Crizotinib-Refractory ALK-Rearranged Non-Small-Cell Lung Cancer: A Phase II Global Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 34, 661-668, doi:10.1200/jco.2015.63.9443 (2016).
31. Crino, L. et al. Multicenter Phase II Study of Whole-Body and Intracranial Activity With Ceritinib in Patients With ALK-Rearranged Non-Small-Cell Lung Cancer Previously Treated With Chemotherapy and Crizotinib: Results From ASCEND-2. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, doi:10.1200/jco.2015.65.5936 (2016).
32. Shaw, A. T. et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. The Lancet. Oncology 18, 1590-1599, doi:10.1016/s1470-2045(17)30680-0 (2017).
33. Kim, D. W. et al. Brigatinib in Patients With Crizotinib-Refractory Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 35, 2490-2498, doi:10.1200/jco.2016.71.5904 (2017).
34. Soria, J. C. et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet (London, England), doi:10.1016/s0140-6736(17)30123-x (2017).
35. Peters, S. et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. The New England journal of medicine, doi:10.1056/NEJMoa1704795 (2017).
36. Camidge, D. R. et al. Brigatinib versus Crizotinib in ALK-Positive Non–Small-Cell Lung Cancer. New England Journal of Medicine 0, null, doi:10.1056/NEJMoa1810171 (2018).
37. Gainor, J. F. et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer discovery, doi:10.1158/2159-8290.cd-16-0596 (2016).
38. Planchard, D. et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. The Lancet. Oncology 17, 984-993, doi:10.1016/s1470-2045(16)30146-2 (2016).
39. Shaw, A. T. et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. The New England journal of medicine 371, 1963-1971, doi:10.1056/NEJMoa1406766 (2014).
40. Li, Z. et al. Efficacy of Crizotinib among Different Types of ROS1 Fusion Partners in Patients with ROS1-Rearranged Non-Small Cell Lung Cancer. J Thorac Oncol 13, 987-995, doi:10.1016/j.jtho.2018.04.016 (2018).
41. Lim, S. M. et al. Open-Label, Multicenter, Phase II Study of Ceritinib in Patients With Non-Small-Cell Lung Cancer Harboring ROS1 Rearrangement. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 35, 2613-2618, doi:10.1200/jco.2016.71.3701 (2017).
42. Gautschi, O. et al. Targeting RET in Patients With RET-Rearranged Lung Cancers: Results From the Global, Multicenter RET Registry. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 35, 1403-1410, doi:10.1200/jco.2016.70.9352 (2017).
43. Awad, M. M. et al. Impact of MET inhibitors on survival among patients (pts) with MET exon 14 mutant (MET del14) non-small cell lung cancer (NSCLC). J Clin Oncol 35, 2017 (suppl; abstr 8511) (2017).
44. Paz-Ares, L. G. et al. PARAMOUNT: Final overall survival results of the phase III study of maintenance pemetrexed versus placebo immediately after induction treatment with pemetrexed plus cisplatin for advanced nonsquamous non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 31, 2895-2902, doi:10.1200/jco.2012.47.1102 (2013).
45. Rittmeyer, A. et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet (London, England) 389, 255-265, doi:10.1016/s0140-6736(16)32517-x (2017).
46. Borghaei, H. et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. The New England journal of medicine 373, 1627-1639, doi:10.1056/NEJMoa1507643 (2015).
47. Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (London, England) 387, 1540-1550, doi:10.1016/s0140-6736(15)01281-7 (2016).
48. Socinski, M. A. et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. The New England journal of medicine, doi:10.1056/NEJMoa1716948 (2018).
49. Kris, M. G. et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. Jama 311, 1998-2006, doi:10.1001/jama.2014.3741 (2014).