Hintergrund
Takotsubo-Syndrom, auch als «Gebrochenes-Herz-Syndrom» bekannt, ist eine Herzerkrankung, die oft durch emotionale oder physische Stressereignisse ausgelöst wird. Die Symptome ähneln denen eines Herzinfarkts. Beim klassischen apikalen Takotsubo-Syndrom gleicht die Ventrikulographie in der Herzkatheteruntersuchung aspektmässig einer Tintenfischfalle, «Takotsubo» auf Japanisch. Stresshormone spielen vermutlich eine Schlüsselrolle. Die Prognose ist im Allgemeinen gut, die meisten Patienten erholen sich innerhalb von Wochen vollständig. Es sind aber auch fatale Verläufe möglich.
Fallbericht
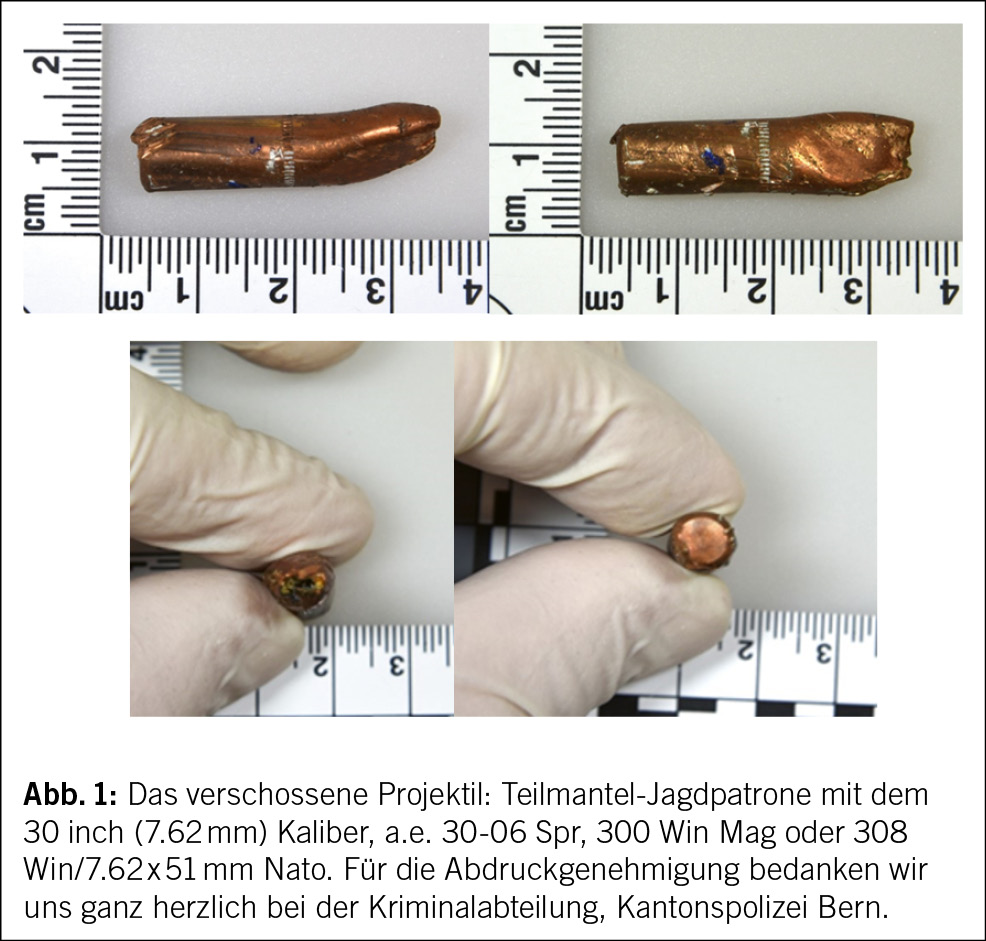
An einem Herbstabend zerbrach ein Projektil das Badezimmerfenster des Einfamilienhauses einer 71-jährigen Frau. Glücklicherweise wurde niemand verletzt. Der Schrecken sass jedoch tief, und die Polizei wurde sofort alarmiert. Im Badezimmer wurde ein Teilmantelgeschoss Kaliber .30 (7.62 mm) aufgefunden (Abb. 1). Mit hoher Wahrscheinlichkeit stammt das Projektil von einer Jagdpatrone. Ermittlungen gegen Unbekannt wurden wegen Gefährdung des Lebens und Sachbeschädigung aufgenommen.
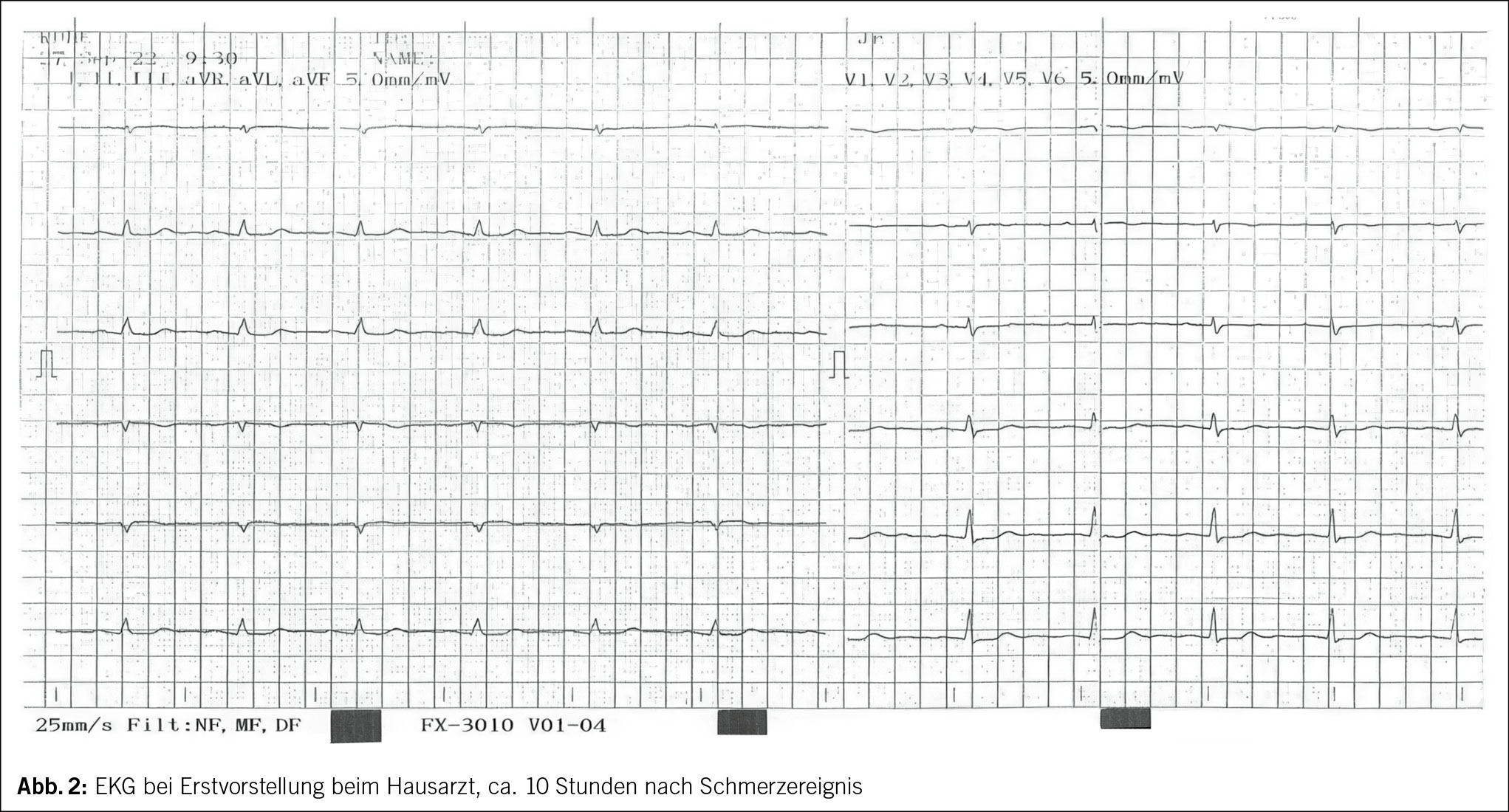
Etwa eine Stunde nach dem Ereignis verspürte die 71-jährige Frau starke Schmerzen in der Brust und zwischen den Schulterblättern, welche nach 1–2 Stunden spontan rückgängig waren. Am Folgetag suchte die Patientin ihren Hausarzt auf, wo ein erhöhtes Troponin Tn-I und anterolaterale sowie inferiore Repolarisationsstörungen mit ST-Senkungen, T-Abflachungen und -Negativierungen im EKG festgestellt wurden (Abb. 2–4).
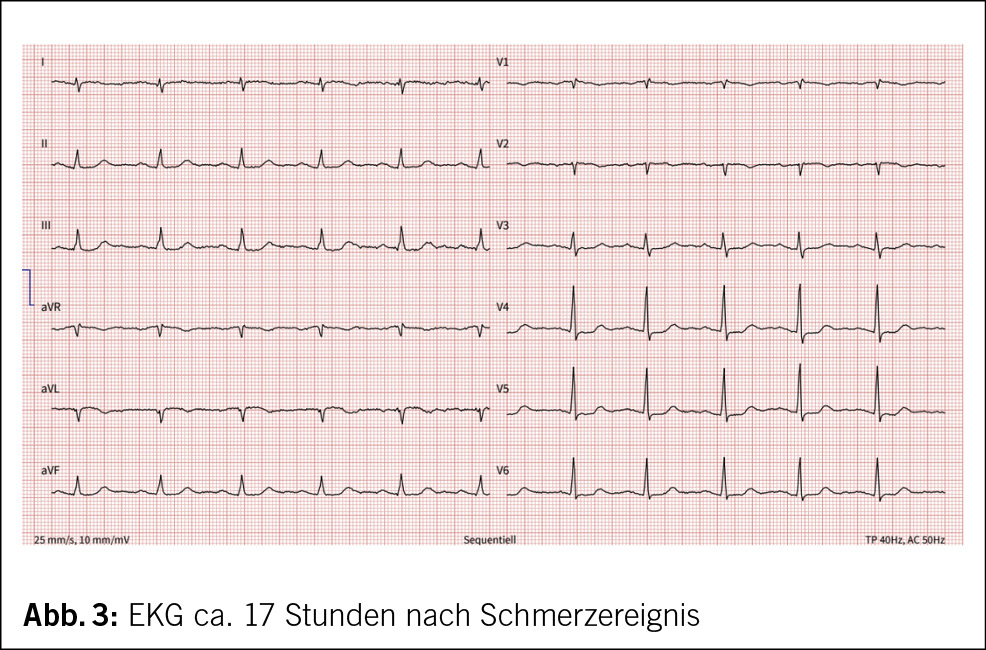
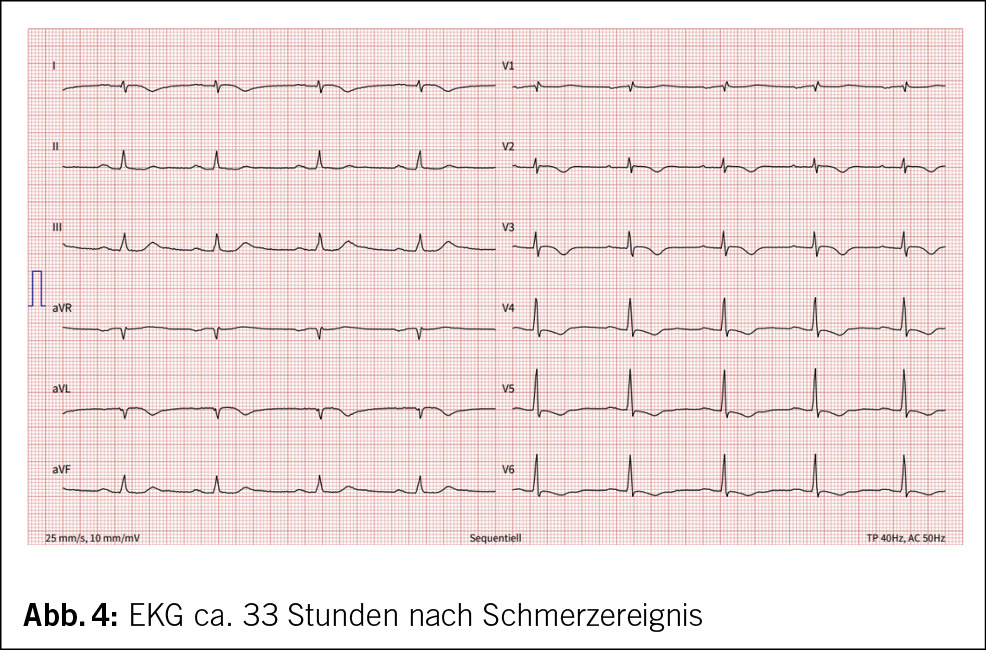
Bei Verdacht auf Myokardinfarkt wurde die Patientin ins regionale Krankenhaus eingewiesen und anschliessend zur Herzkatheteruntersuchung ins Zentrumsspital verlegt. Eine koronare Herzkrankheit konnte ausgeschlossen werden, jedoch zeigte die Ventrikulographie bei einem erhöhten linksventrikulären enddiastolischen Druck vom 20 mmHg (Norm < 15 mmHg) eine schwer eingeschränkte systolische linksventrikuläre Ejektionsfraktion (LVEF) von 25 % (Norm > 55 %) mit Akinesie der midventrikulären Abschnitte (Abb. 5).
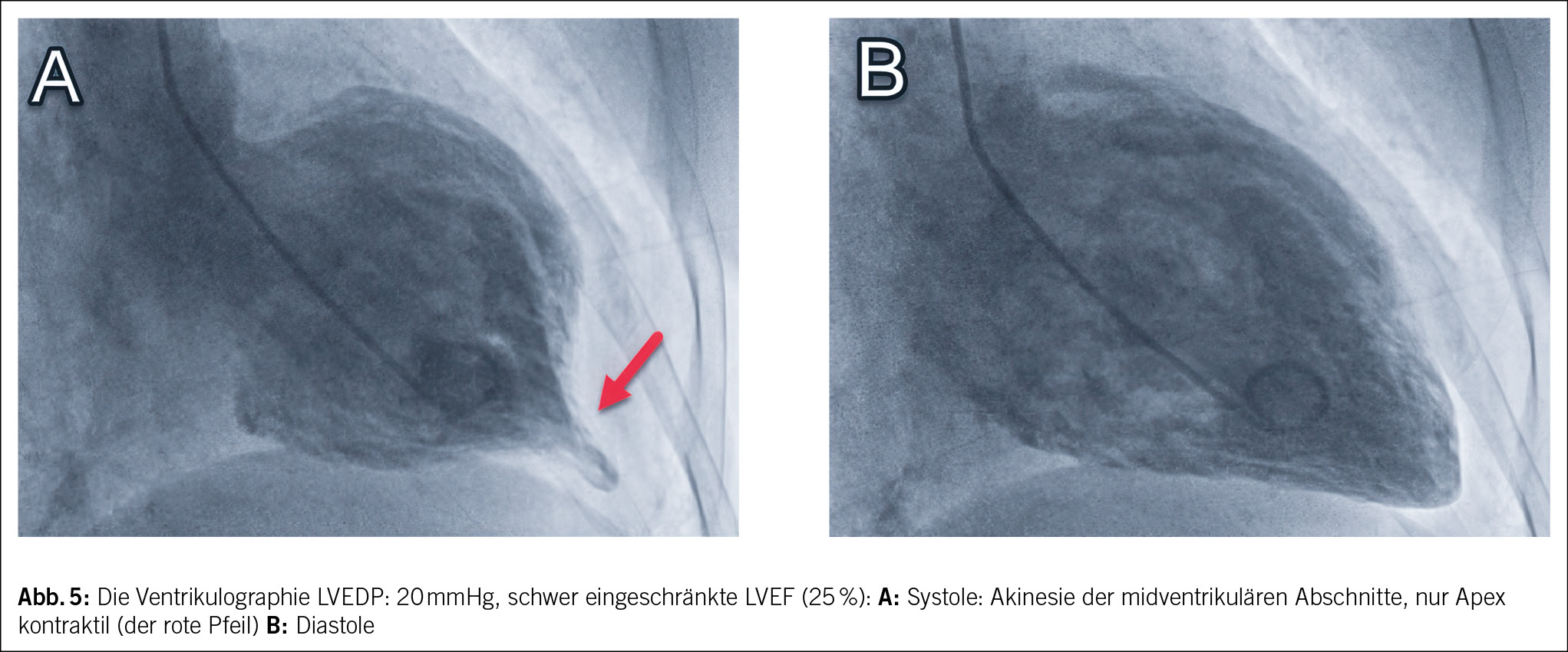
Die Diagnose eines midventrikulären Takotsubo-Syndroms wurde gestellt. Es wurde anschliessend eine Herzinsuffizienztherapie mit Betablocker, ACE-Hemmer, SGLT-2-Inhibitor, Spironolacton und Schleifendiuretika begonnen. Bereits am dritten Hospitalisationstag zeigte die transthorakale echokardiographische Kontrolle eine deutliche Besserung der LVEF von 25 % auf 45 %. Nach 4 Tagen konnte die Patientin aus der Spitalpflege entlassen werden. 3 Monate später hatte sich die LVEF vollständig normalisiert, die Patientin war beschwerdefrei, und die Herzinsuffizienztherapie wurde bis auf das Sartan (zwischenzeitlich Wechsel erfolgt) gestoppt. Aus der Vorgeschichte der Patientin ist bekannt, dass sie bereits früher ähnliche Brustschmerzen erlebte, die mit dem emotionalen Ereignis im Zusammenhang standen, als ihr Sohn ins Ausland auswanderte. Es erfolgte damals jedoch keine eingehende kardiale Untersuchung.
Diskussion
Definition
Das Takotsubo-Syndrom ist eine transiente LV-Dysfunktion mit typischen Wandbewegungsstörungen, welche nicht auf eine koronare Stenose oder einen Verschluss zurückzuführen sind, häufig von physischen oder emotionalen Triggern ausgehend (1).
Symptomatik und Diagnostik
Das Krankheitsbild des Takotsubo-Syndroms ist durch typische Beschwerden ähnlich eines akuten Koronarsyndroms mit akuten thorakalen Schmerzen, Dyspnoe sowie erhöhten kardialen Biomarkern gekennzeichnet. Elektrokardiographisch zeigen sich typischerweise ST-Strecken-Abnormalitäten, am häufigsten initial ST-Hebungen und im Verlauf fortschreitende T-Inversionen und QT-Zeit-Verlängerungen (2, 3). Echokardiographisch und in der Ventrikulographie sind typische regionale Wandbewegungsstörungen sichtbar. Die Koronararterien zeigen typischerweise keinen erklärenden Koronarverschluss. Anhand der Regionalitäten lassen sich morphologisch 4 Typen des Takotsubo-Syndroms unterscheiden: den midventrikulären Typ, den basalen und den fokalen Typ sowie den apikalen Typ. Letzterer weist als typisches Bild ein apikales Ballooning infolge der apikalen Akinesie auf und tritt in über 80 % aller Fälle auf (4). Der midventrikuläre Typ ist die zweithäufigste Form, dabei sind die midventrikulären Wandabschnitte akinetisch und die basalen sowie apikalen Segmente hyperkontraktil. Gemäss dem internationalen Takotsubo-Register wird vermutet, dass die LVEF bei atypischem Takotsubo-Syndrom im Vergleich mit dem typischen Takotsubo-Syndrom weniger eingeschränkt ist (5).
Wichtig für die Praxis
Da ein Takotsubo-Syndrom nicht invasiv nicht eindeutig von einem akuten Koronarsyndrom unterschieden werden kann, ist die initiale Behandlung identisch mit der eines akuten Koronarsyndroms, und der wichtigste diagnostische Schritt ist eine Linksherzkatheteruntersuchung.
Trigger und Demographie
Typische Auslöser für Takotsubo-Syndrom sind physische und psychische Trigger unterschiedlicher Ausprägung. Bei den psychischen Triggern handelt es sich um negative oder positive emotionale Ereignisse. Beispiele für positive Trigger sind ein Lottogewinn, für negative Trigger der Tod einer nahestehenden Person, eine Scheidung, finanzielle Probleme, ein Erdbeben, Krieg (6, 7) oder auch Angst vor der COVID-19-Pandemie (8). In etwas über einem Viertel aller Fälle findet man jedoch keine Auslöser (9). Der Grossteil der Betroffenen sind Frauen mit einem mittleren Alter von 66 Jahren (4).
Pathomechanismus
Der genaue Mechanismus ist nicht eindeutig. Es sprechen viele Studien für eine akute mikrovaskuläre Dysfunktion als Hauptursache des Takotsubo-Syndroms (10). Jedoch sind auch direkte Effekte auf die Kardiomyozyten nachweisbar: Während Stresssituationen kommt es zur Ausschüttung von Katecholaminen. Hohe Dosen von Epinephrin führen zu einer Signaltransduktion (11). Bei niedriger Epinephrinspiegel werden über den β2-Rezeptor stimulierende G-Proteine aktiviert, was positiv inotrop wirkt. Im Falle hoher Epinephrinspiegel werden anstatt stimulierender G-Proteine inhibitorische G-Proteine (Gi) aktiviert, was negativ inotrop wirkt. Es wird angenommen, dass die β2-Rezeptordichte im linken Ventrikel apikal höher ist als basal, was das apikale Ballooning erklären kann (12). Die Patienten mit einem Takotsubo-Syndrom-Rezidiv können eine andere Form zeigen als die Primärmanifestation (13). Es kann durch die Down-Regulation der β2-Rezeptoren nach erster sympathischer Stimulation erklärt werden (14–15). Als weitere Ursachen werden Gefässspasmen und endotheliale Dysfunktionen mit Mikrozirkulationsproblemen diskutiert (1).
Therapie
Die Empfehlungen beruhen auf retrospektiven Studien sowie Expertenmeinungen (12).
Als Komplikationen können eine akute Herzinsuffizienz oder Rhythmusstörungen auftreten. In der Regel richtet sich die Behandlung nach den Leitlinien der einzelnen Krankheit (z. B. ventrikuläre Tachykardie oder akute Herzinsuffizienz). Somit werden meist eine Herzinsuffizienztherapie und eine rhythmologische Überwachung durchgeführt. Es wird empfohlen, die rhythmologische Überwachung bis zur Normalisierung der QT-Zeit durchzuführen (3).
Falls ein kardiogener Schock auftritt, muss eine dynamische Obstruktion des linksventrikulären Ausflusstraktes (LVOT-Obstruktion) echokardiographisch evaluiert werden: Bei einem Takotsubo-Syndrom des apikalen Typs bleibt oft als einziger kontraktiler Teil des Myokards die basale Manschette, dazu gehört auch die septale Wand des LVOT. Dieser kompensatorisch hyperkontraktile Wandabschnitt des LVOT behindert den Auswurf in die Aorta direkt. Zudem kommt es durch die Flussbeschleunigung im engen LVOT zu einem systolischen Ansaugen des anterioren Mitralsegels (SAM), was die Obstruktion verstärkt. Eine zusätzliche Folge ist die schwere Mitralklappeninsuffizienz, welche das Herzminutenvolumen nochmals reduziert. Inotropika können so schlussendlich zu einem akuten Pumpversagen führen. Die richtige Behandlung gleicht der einer hypertrophen obstruktiven Kardiomyopathie mit Erhöhung der Nachlast (z. B. Noradrenalin) sowie Betablockade und Volumentherapie. Falls keine Stabilisierung erfolgt, kann Impella erwogen werden (2). Die LVOT-Obstruktion tritt bei 10–25 % von allen Fällen auf (16).
Falls keine LVOT-Obstruktion auftritt, unterscheidet sich die Therapie nicht grundsätzlich von der einer akuten Herzinsuffizienz anderer Ursache. Bei einer LVEF < 30 % sollte die Antikoagulation in Betracht gezogen werden. Die Therapie mit ACE-Hemmern/Angiotensin-II-Rezeptorblockern ist mindestens bis zur Normalisierung der LVEF empfohlen. Jedoch zeigen retrospektive Analysen einen Benefit hinsichtlich Mortalität und Rezidivrate unter ACE-Hemmern, weshalb eine Dauertherapie evaluiert werden sollte. Die langfristige Therapie mit Betablockern ergab interessanterweise trotz der Pathophysiologie keine Vorteile (3), die 30-Tage-Sterblichkeit ist nicht reduziert (17).
Prognose
Auch wenn sich die meisten Patienten vollständig von einem Takotsubo-Syndrom erholen, kann das Krankheitsbild nicht als gutartig bezeichnet werden. In der akuten Phase tritt in 5–10 % aller Fälle ein kardiogener Schock auf. Anhand der International Takotsubo Registry Study liegt die Mortalität bei 4.1 %, das Takotsubo-Syndrom hat also eine vergleichbare Komplikations- und Mortalitätsrate wie ein akutes Koronarsyndrom (4).
Falls die akute Phase überlebt wird, normalisiert sich die systolische linksventrikuläre Ejektionsfraktion in der Regel innerhalb von 1 bis 4 Wochen (18). Die Symptome wie Dyspnoe, Lethargie, Herzrasen und Brustschmerz können sogar mehr als 2 Jahre nach dem Ereignis persistieren, trotz Normalisierung der LVEF (8). Das jährliche Risiko, ein Rezidiv zu erleiden, liegt bei 1–2 % pro Jahr (4). In den ersten 5 Jahren tritt ein Rezidiv bei einem von acht Patienten auf, wobei der Auslöser meistens anders ist als beim ersten Ereignis (8).
Zusammenfassung
Bei unserem Fall hat das Gewehrprojektil die Patientin glücklicherweise nicht getroffen. Der Vorfall war trotzdem für die Patientin ein aussergewöhnliches emotionales Ereignis. Durch Ausschüttung von Stresshormonen hat der Vorfall aber seine indirekte und potenziell tödliche Wirkung auf das Herz entfaltet. Es entwickelte sich ein Takotsubo-Syndrom mit den typischen Thoraxschmerzen, Repolarisationsstörungen sowie positiver Dynamik der kardialen Biomarker. Atypisch war die Morphologie mit midventrikulärer Wandbewegungsstörung. In unserem Fall ist eine frühere Episode eines Takotsubo-Syndroms zu erahnen. Erfreulicherweise war der Verlauf benigne, und die systolische LVEF hat sich nach 3 Monaten unter ausgebauter Herzinsuffizienztherapie normalisiert. Die gefürchteten Komplikationen einer LVOT-Obstruktion sind nicht aufgetreten. Es ist nicht der erste Fallbeschrieb eines Takotsubo-Syndroms nach einer Schussabgabe, jedoch der erste ohne direkte Wirkung durch das Projektil.
Verdankung(en)
Wir bedanken uns für die Abdruckgenehmigung sowie die Fallinformationen bei der Kriminalabteilung, Kantonspolizei Bern, sowie der Staatsanwaltschaft des Kantons Bern.
Ethics Statement
Ein schriftlicher Informed Consent zur Publikation liegt vor.
Author Contributions
Gleicher Anteil aller Autoren
Martina Oslayová
Christoph Gräni
Christian Muster
Universitätsklinik für Kardiologie, Inselspital Bern
Oberarzt Kardiologie
Universitätsklinik für Kardiologie
Inselspital Bern
Freiburgstrasse 20
3010 Bern
christian.muster@insel.ch
Die Autorinnen und Autoren haben deklariert, keine potenziellen Interessenkonflikte zu haben.
1. International Expert Consensus Document on Takotsubo Syndrome (Part I): Clinical Characteristics, Diagnostic Criteria and Pathophysiology, J.-R. Ghadri et al. European Heart Journal (2018) 39, 2032–2046
2. Parkkonen O, Allonen J, Vaara S, Viitasalo M, Nieminen MS, Sinisalo J, Differences in ST-elevation and T-wave amplitudes do not reliably differentiate takotsubo cardiomyopathy from acute anterior myocardial infarction., J Electrocardiol. 2014 Sep-Oct;47(5):692-9. Epub 2014 Jun 14.
3. J.-R. Ghadri et al., International Expert Consensus Document on Takotsubo Syndrome (Part II): Diagnostic Workup,Outcome, and Management, European Heart Journal (2018) 39, 2047–2062
4. Templin C, Ghadri JR, Diekmann J, et al. Clinical Features and Outcomes of Takotsubo (Stress) Cardiomyopathy. N Engl J Med 2015; 373: 929–38.
5. Ghadri JR, Cammann VL, Napp LC, Jurisic S, Diekmann J, Bataiosu DR, Seifert B, Jaguszewski M, Sarcon A, Neumann CA, Geyer V, Prasad A, Bax JJ, Ruschitzka F, Luscher TF, Templin C; International Takotsubo Registry. Differences in the clinical profile and outcomes of typical and atypical takotsubo syndrome: data from the International Takotsubo Registry. JAMA Cardiol 2016;1: 335–340
6. Schlossbauer et al., Takotsubo-Syndrom – ein häufig verkanntes Krankheitsbild. Praxis 2016, 1185-1192
7. Rikabi et al., Takotsubo Cardiomyopathy Triggered by Emotional Stress From the Russia-Ukraine War, JACC: CASE REPORTS, VOL. 16, JUNE 21, 2023
8. Trisha Singh et al., Takotsubo Syndrome: Pathophysiology, Emerging Concepts, and Clinical Implications, Circulation. 2022;145:1002–1019
9. Pelliccia F et al., Comorbidities frequency in Takotsubo syndrome: an international collaborative systematic review including 1109 patients. Am J Med. 2015;128(6):654.e11. Epub 2015 Feb 4.
10. Galiuto et al. Reversible coronary microvascular dysfunction: a common pathogenetic mechanism in Apical Ballooning or Tako-Tsubo Syndrome. European Heart Journal (2010), 1319–1327
11. Paur H, Wright PT, Sikkel MB et al (2012) High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/Gi-dependent manner: a new model of Takotsubo cardiomyopathy. Circulation 126:697–706
12. L. Christian Napp und Johann Bauersachs, Takotsubo-Kardiomyopathie Springer Medizin, 29.07.2015
13. Xu B, Williams PD, Brown M, Macisaac A (2014) Takotsubo cardiomyopathy: does recurrence tend to occur in a previously unaffected ventricular wall region? Circulation 129(7):e339–e340
14. Lyon AR, Citro R, Schneider B, et al. Pathophysiology of takotsubo syndrome: JACC state-of-the- art review. J AmColl Cardiol. 2021;77:902–921.
15. El-Battrawy I, Santoro F, Stiermaier T, et al. Incidence and clinical impact of recurrent takotsubo syndrome: results from the GEIST Registry. J Am Heart Assoc. 2019;8:e010753.
16. Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004, 141:858.
17. Isogai t., Matsui H., Tanaka H. et al., Early Betablocker use and in-hospital mortality in patients with Takotsubo cardiomyopathy. Heart 2016, 102:1029
18. Sharkey SW, Lesser JR, Zenovich AG, et al. Acute ans reversible cardiomyopathy provoked by stress in women from the United States. Circulation 2005, 111:472