Fallbericht
Ein 79-jähriger Patient wurde aufgrund einer fortgeschrittenen, symptomatischen Coxarthrose zur elektiven Implantation einer Hüftprothese in unser Spital aufgenommen. An relevanten Vorerkrankungen lagen eine arterielle Hypertonie, eine Hypercholesterinämie, ein Vorhofflimmern sowie ein Status nach ischämischem Kleinhirninfarkt links vor zwei Jahren vor. Die Dauermedikation bestand aus Rivaroxaban 20 mg, Valsartan 160 mg und Rosuvastatin 20 mg.
Die Operation verlief am Aufnahmetag planmässig. Dem Patienten wurde eine zementierte Hüfttotalprothese nach vorherigem Einsetzen einer Markraumsperre implantiert.
Unmittelbar nach dem Eingriff zeigte sich der Patient wach, orientiert und gab abgesehen von Hüftschmerzen keine Beschwerden an. In der folgenden Nacht fiel jedoch eine psychomotorische Verlangsamung auf, die weitere neurologische Untersuchung zeigte zudem eine Desorientierung, eine Dysarthrie und ein Absinken im Armhalteversuch rechts (NIHSS: 8 Punkte). Die Vitalparameter waren mit einem Blutdruck von 132/79 mmHg sowie einer Pulsfrequenz von 92/min im Normbereich. Die Sauerstoffsättigung betrug minimal 91 %. In der internistischen Untersuchung fanden sich an den Händen subunguale Hämorrhagien bei sonst unauffälligen Befunden.
Laborchemisch zeigte sich eine Anämie von 9.3g/dl sowie eine milde Thrombozytopenie von 142 × 10⁹/l. Der Blutzucker war mit 5.4 mmol/l normwertig.
Als Ursache der akuten neurologischen Symptome wurde primär ein Schlaganfall bei bekanntem Vorhofflimmern und pausierter oraler Antikoagulation vermutet. Differenzialdiagnostisch standen unter anderem protrahierte medikamentöse Nebenwirkungen nach Vollnarkose oder ein postiktaler Zustand nach möglichem unbeobachtetem Anfall zur Diskussion (Tab. 1).
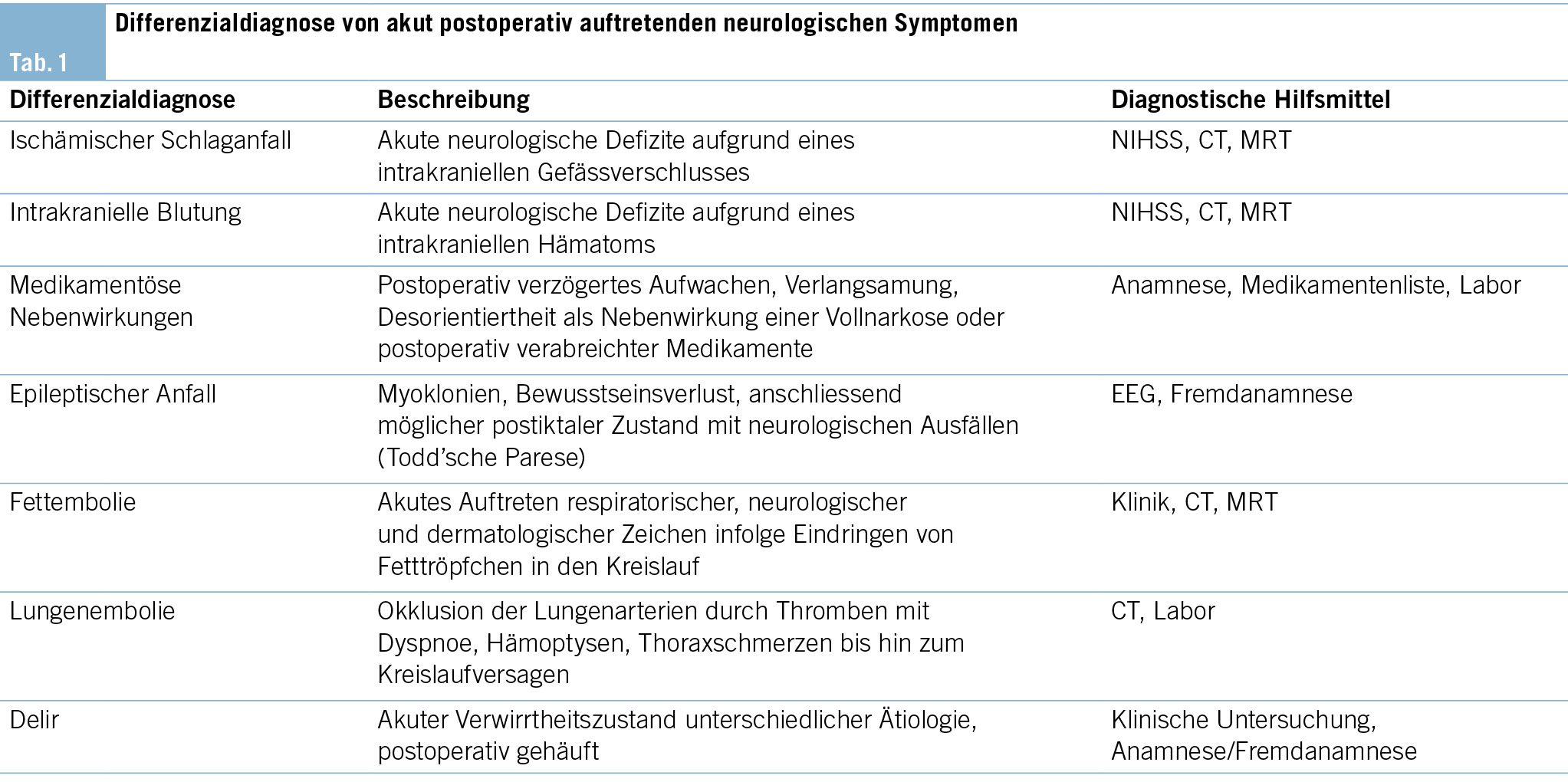
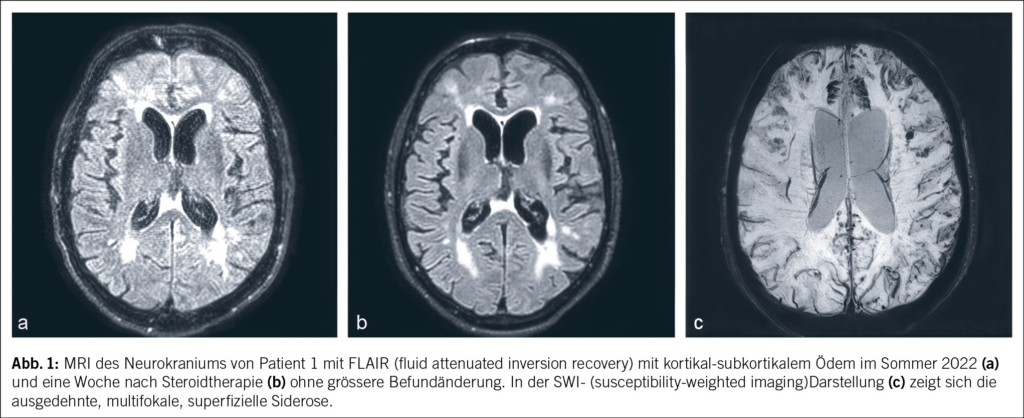
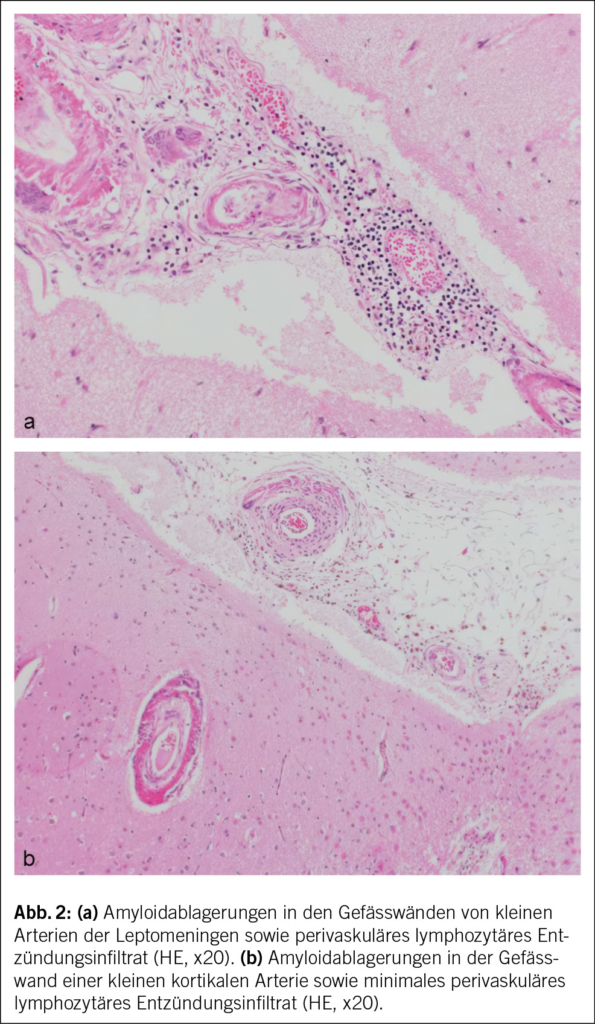
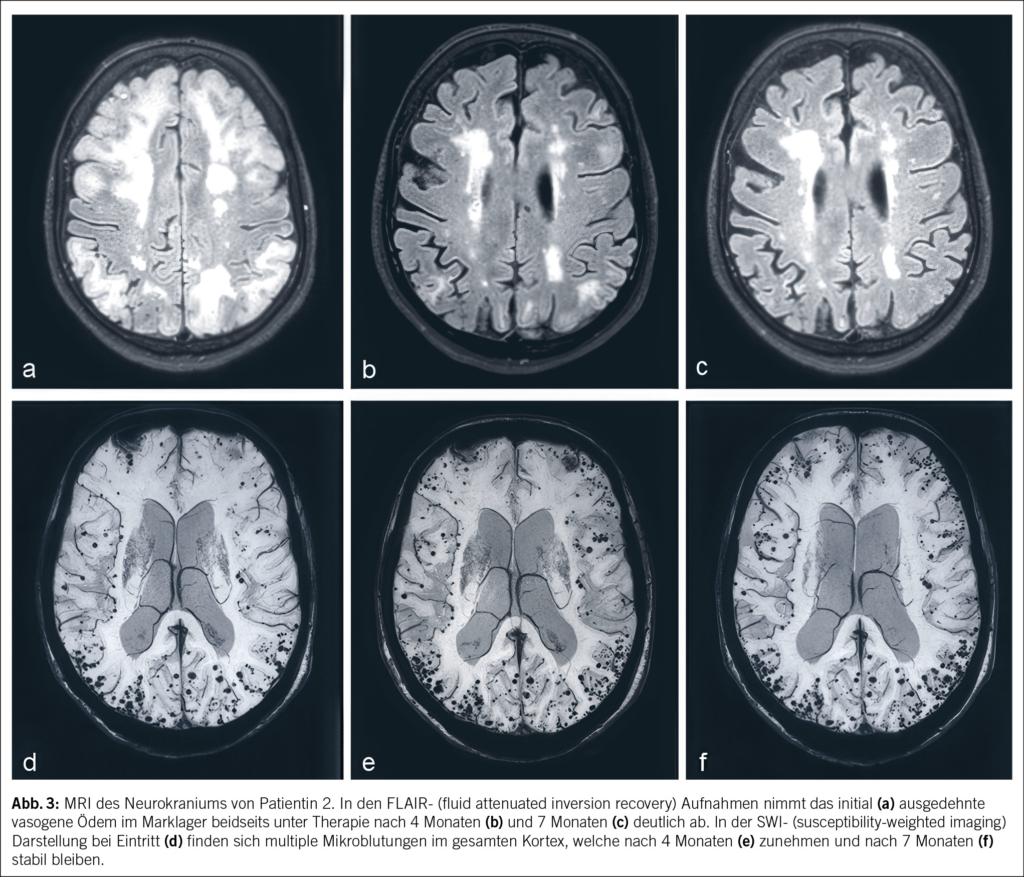
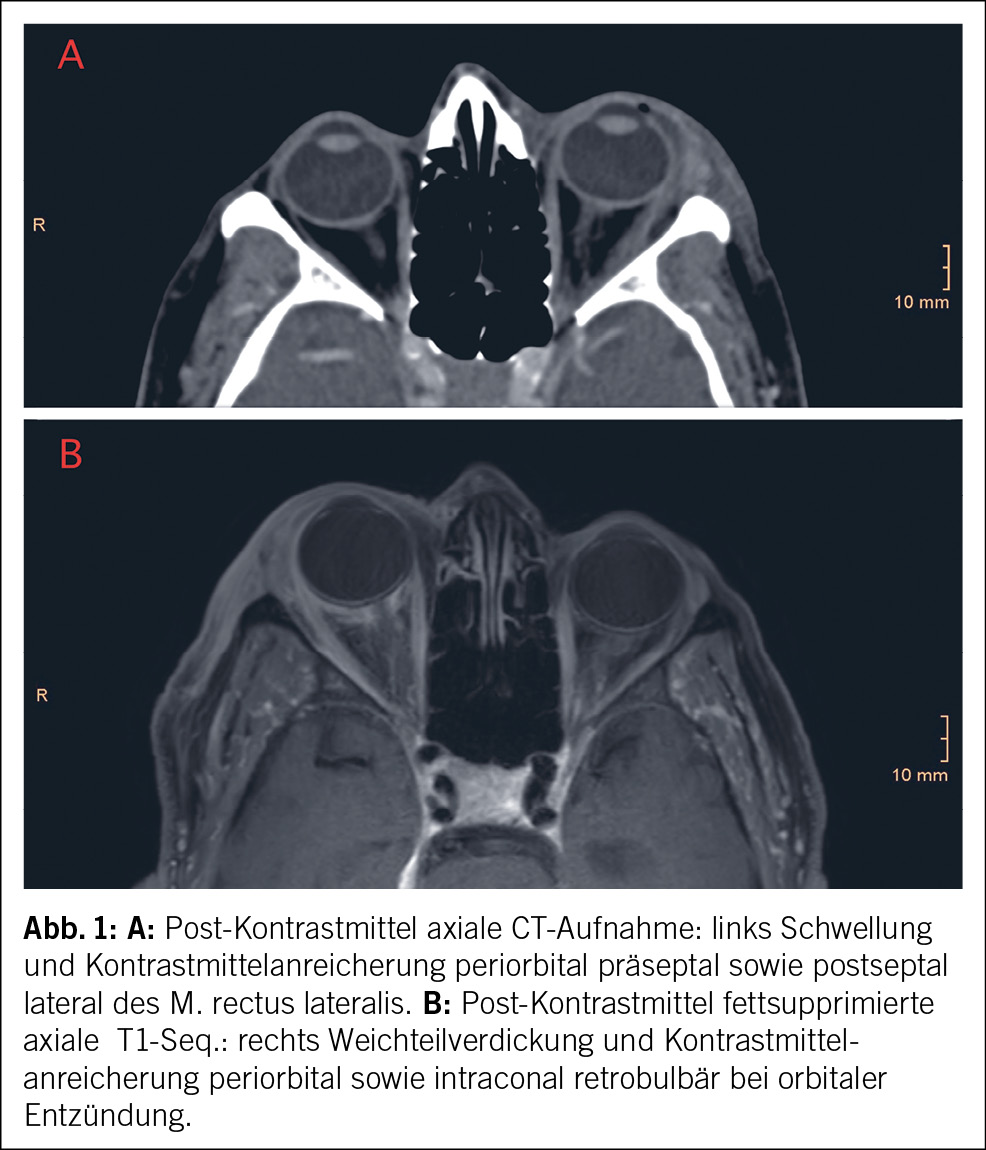
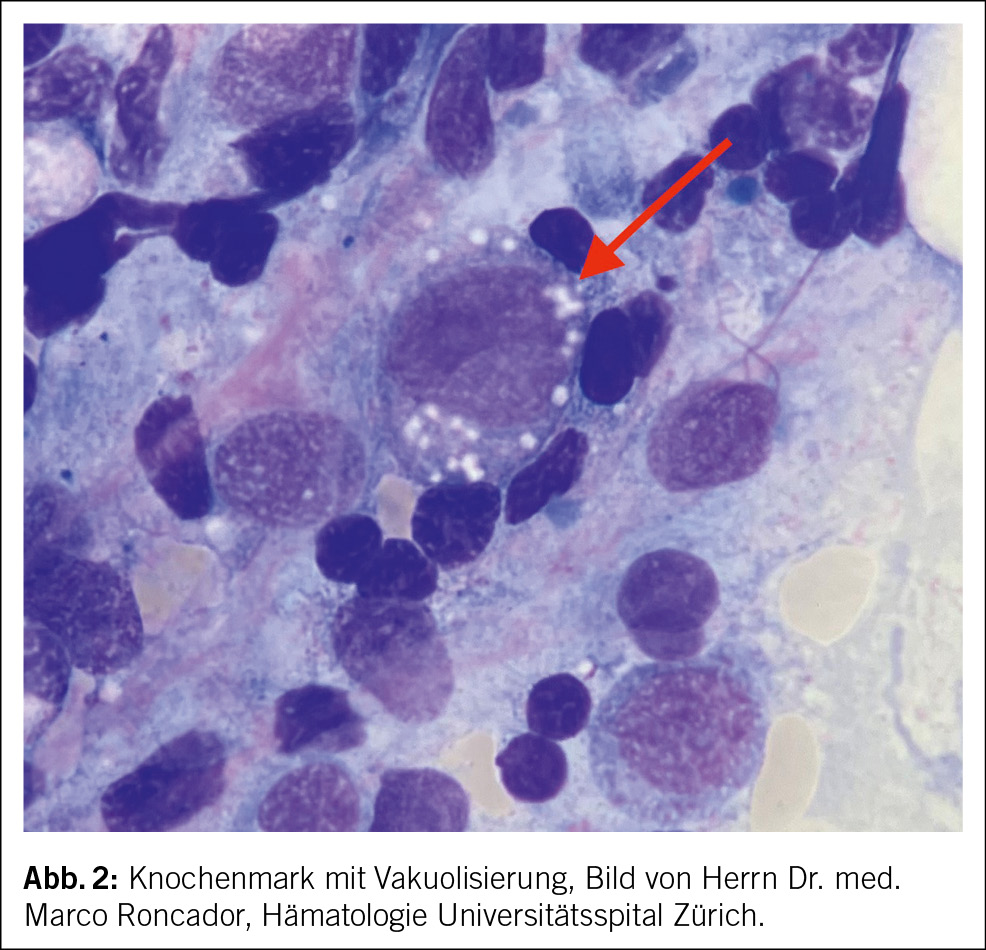
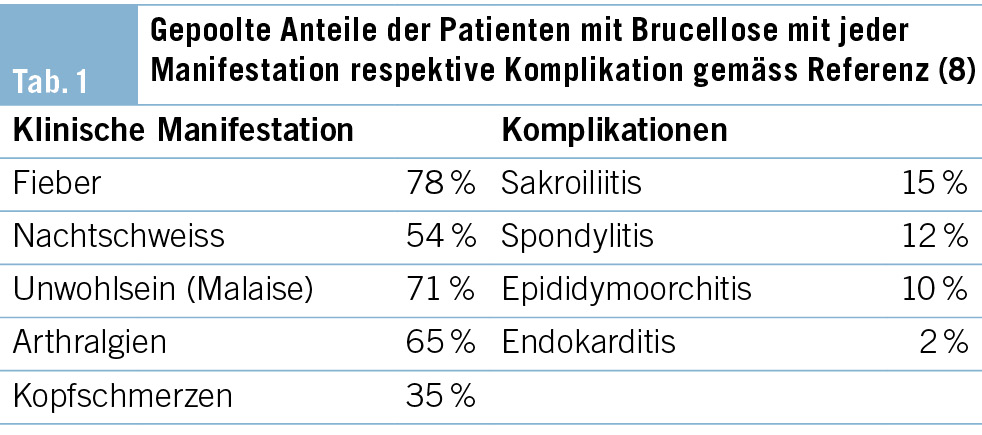
In der notfallmässigen Schädel-CT mit Angiographie und Perfusion zeigte sich ein normaler Befund ohne Hinweise auf eine intrakranielle Blutung, eine frische Ischämie oder einen Gefässverschluss. Am Folgetag wurde eine erweiterte Diagnostik mit MRT des Kopfes und EEG durchgeführt. Letzteres zeigte ein enzephalopathisches Bild ohne epilepsietypische Potenziale. In der MRT konnten diffuse, im Grosshirn verteilte, punktförmige Ischämien wie auch Mikroblutungen nachgewiesen werden (Abb. 1 und Abb. 2). Aufgrund des charakteristischen radiologischen Bildes («starfield pattern») ergab sich der Verdacht auf eine zerebrale Fettembolie. Aufgrund negativer Duke-Kriterien galt eine aufgrund des MRT-Befundes differenzialdiagnostisch evaluierte Endokarditis als unwahrscheinlich. Eine Thorax-CT mit Angiographie schloss eine Lungenembolie oder anderweitige pulmonale Auffälligkeiten aus.
Es erfolgte eine rein supportive Therapie. Abgesehen von einer intensiven physio- und ergotherapeutischen sowie logopädischen Betreuung wurden keine weiteren spezifischen Massnahmen ergriffen. Im Verlauf der nächsten 14 Tage kam es darunter zu einer kompletten Rückbildung der neurologischen Symptome. Am zweiten postoperativen Tag wurde eine prophylaktische Antikoagulation mittels Rivaroxaban 10 mg begonnen. Die ursprüngliche Dosierung von 20 mg wurde am 15. postoperativen Tag wiederaufgenommen.
Diskussion
Eine Fettembolie (FE) beschreibt den Verschluss kleiner Gefässe durch im Blut transportierte Fettpartikel. Dabei sind insbesondere die Präkapillaren und Kapillaren der Lunge betroffen. Gelangen einzelne Fettpartikel durch die Lunge in den arteriellen Kreislauf und verschliessen dort die kleineren Gefässe, etwa in der Niere, der Haut oder im Gehirn, spricht man von einer arteriellen FE. Treten klinische Symptome auf, wird dies als Fettembolie-Syndrom (FES) bezeichnet (1).
Das FES, erstmalig 1861 beschrieben, besteht klassischerweise aus der Trias von akuter respiratorischer Insuffizienz, zerebraler Dysfunktion und Petechien. Hauptsächlich tritt das FES im Zusammenhang mit Frakturen langer Knochen, insbesondere des Femurs oder des Beckens, auf.
Die Inzidenz einer asymptomatischen FE liegt bei Traumapatienten bei bis zu 90 %, während die Inzidenz eines FES bei Patienten mit einer Fraktur eines langen Knochens zwischen 0.5 und 2 % variiert. Bei multiplen Knochenfrakturen steigt sie auf 5–10 %. Die Mortalität liegt in diesen Fällen zwischen 1 und 20 % (1, 2, 3), wobei bei Patienten, welche Symptome innerhalb der ersten zwölf Stunden nach dem auslösenden Ereignis entwickeln, eine deutlich höhere Mortalität besteht. Ein milderer Verlauf wird entsprechend beschrieben, wenn sich die Symptome erst nach 24 bis 72 Stunden entwickeln (3).
Die Pathogenese der Fettembolisation bzw. des Fettembolie-Syndroms ist nicht vollständig geklärt. Es bestehen drei pathophysiologische Theorien, wobei auch eine Kombination daraus möglich ist:
1. Die mechanische Theorie postuliert, dass anlässlich eines Knochentraumas Fett aus dem Knochenmark in den Blutkreislauf übertritt. Bei einem Trauma grosser Knochen mit höherem Knochenmarkanteil kann es zu Verletzungen oder Abrissen der am Knochen anliegenden Venen kommen. Dadurch können Knochenmarkbestandteile leichter in den Blutkreislauf eindringen. Während orthopädischer Operationen können die Manipulation der Knochen oder das Einbringen von Marknägeln oder Markraumsperren den intramedullären Druck erhöhen, wodurch das Eindringen von intramedullärem Fett in den Kreislauf begünstigt wird.
2. Gemäss einer biochemischen Hypothese kommt es nach einem Trauma zu einer Freisetzung von freien Fettsäuren in Form von Chylomikronen, Low Density Lipoproteinen (LDL) und Liposomen. Durch eine Agglutination mit Akute-Phase-Proteinen kann es zu einer Embolisierung kommen (4, 12).
3. Eine weitere These geht davon aus, dass es bei Frakturen grosser Knochen zu einer Freisetzung von Thromboplastin kommt, wodurch die Aktivierung des Komplementsystems sowie der extrinsischen Koagulationskaskade ausgelöst wird (6).
Eine Fettembolie kann durch Obstruktion pulmonaler Kapillaren respiratorische Symptome hervorrufen. Ist sie ausgeprägt, führt ein Ventilations-Perfusions-Mismatch zu einer ausgeprägten Hypoxämie, welche ein schweres Rechtsherzversagen zur Folge haben kann (4). Darüber hinaus kann eine FE durch eine paradoxe Embolie (z.B. über ein offenes Foramen ovale) oder eine Mikroembolisation in den arteriellen Kreislauf gelangen (5). In einem solchen Fall können neurologische Symptome und Petechien auftreten. Die neurologischen Symptome entstehen vermutlich durch zwei Mechanismen: durch die Obstruktion kleiner cerebraler Arteriolen und durch die zytotoxische Wirkung freigesetzter Fettsäuren, welche eine erhöhte kapilläre Permeabilität und damit einhergehend zerebrale Mikroinfarkte und Mikroblutungen verursachen (6).
Typischerweise manifestiert sich ein FES 24–72 Stunden nach dem auslösenden Trauma. Respiratorische Symptome, beispielsweise Dyspnoe, Tachypnoe und Hypoxämie, sind häufig die ersten klinischen Anzeichen. Eine Hypoxämie tritt bei bis zu 96 % der betroffenen Patienten auf (3). Zusätzlich können neurologische Ausfälle auftreten.
bis hin zu schweren fokalen Ausfällen (7). Im Falle einer primär neurologischen Symptomatik wird von einer zerebralen Fettembolie (ZFE) gesprochen – einer seltenen Form, die etwa 10 % der FE-Fälle ausmacht (8). In einem Drittel der Fälle treten zudem Petechien auf, die durch eine Embolisation kleinerer Hautgefässe ausgelöst werden. Petechien zeigen sich typischerweise 36 Stunden nach dem auslösenden Ereignis und sind nach sieben Tagen in der Regel nicht mehr nachweisbar. Unspezifische Manifestationen des FES können ferner Fieber, Tachykardie oder laborchemische Auffälligkeiten wie eine Thrombozytopenie oder Anämie sein. In den meisten Fällen ist ein FES innerhalb weniger Tage vollständig reversibel (6).
Die Diagnose des FES wird primär klinisch gestellt. Es existieren verschiedene Diagnosekriterien, welche allerdings nicht validiert oder universell akzeptiert sind (9) (10). Die Gurd-Kriterien, die später von Wilson modifiziert wurden, werden am häufigsten verwendet (Tab. 2). Für die Diagnose eines FES müssen entweder zwei Hauptkriterien oder ein Haupt- und vier Nebenkriterien erfüllt sein (7, 11). Obwohl die Diagnose primär klinisch gestellt wird, können bildgebende Verfahren, insbesondere beim ZFE, eine wertvolle Unterstützung bieten.

fusionsgewichteten Sequenzen der Nachweis eines «starfield pattern», welches durch multiple kleine, nicht konfluierende, hyperintense Läsionen gekennzeichnet ist (Abb. 1)(12). Das «starfield pattern», erstmalig 2001 von Parizel et al. beschrieben, ist als unspezifisch charakterisiert und ein Zeichen von diffusen, subkortikalen Mikroembolien (13). Ein wichtiges Zusatzkriterium sind subkortikale Mikroblutungen in den SWI- und T2-Sequenzen, welche ebenfalls diffus verteilt auftreten und eine einheitliche Grösse haben (Abb. 2). Typischerweise liegen diese in einem «walnut kernel pattern» vor (14). Laborchemische Untersuchungen sowie Sputum- oder Urinanalysen zum Nachweis von Fettpartikeln gelten hingegen als unspezifisch. Da die oben genannten diagnostischen Kriterien vor der Einführung der MRT entwickelt wurden, ist die MRT in den bisherigen Kriterien noch nicht als wichtiges diagnostisches Mittel verankert.
Die Therapie des FES erfolgt symptomorientiert. Im Vordergrund stehen eine adäquate Oxygenierung, Stabilisierung der Hämodynamik und Volumensubstitution (7, 12). Als mögliche weitere Therapie wird der Einsatz von Kortikosteroiden diskutiert, welche durch eine Stabilisierung der pulmonalen Kapillarmembran, die Unterbindung einer Entzündungsreaktion und die Verzögerung einer Plättchenadhäsion ein FES verhindern sollen. Eine Metaanalyse, die 389 Patienten aus sechs Studien einschloss, zeigte, dass die Gabe von Kortikosteroiden das Risiko eines FES um 80 % reduzieren konnte. Einflüsse auf die Mortalität konnten hingegen nicht nachgewiesen werden (1, 15). Die Anwendung von Kortikosteroiden ist weiterhin umstritten, da sie insbesondere die Erhöhung des Infektionsrisikos bedingen kann (16). Sonstige medikamentöse Massnahmen, etwa die Gabe von Heparin, Albumin oder Aspirin, zeigten in den Studien keinen nachweisbaren Nutzen.
Wichtige präventive Massnahmen zur Vermeidung eines FES umfassen die frühzeitige Reponierung von Frakturen sowie die Begrenzung des intraossären Drucks während orthopädischer Eingriffe.
Zusammenfassend hat es sich bei dem beschriebenen Patientenfall um eine isolierte zerebrale Fettembolie gehandelt, da die Kriterien für eine FES letztlich nicht eindeutig erfüllt waren. Die Diagnose der ZFE wurde im Hinblick auf den zeitlichen Zusammenhang mit der OP sowie der MRT-Bildgebung gestellt.
Abkürzungen:
CT Computertomographie
EEG Elektroenzephalogramm
FE Fettembolie
FES Fettembolie-Syndrom
LDL Low Density Lipoprotein
MRT Magnetresonanztomographie
NIHSS National Institutes of Health Stroke Scale
ZFE Zerebrale Fettembolie
Historie
Manuskript eingegangen: 05.11.2024
Angenommen nach Revision: 22.01.2025
Klinik für Allgemeine Innere Medizin
Spitäler Schaffhausen
Geissbergstrasse 81
8208 Schaffhausen
Die Autorenschaft hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. C. Forster, M. Jöhr und J.-O. Gebbers, «Fettembolie und Fettembolie-Syndrom,» Schweiz Med Forum Nr. 28, pp. 673-678, 2002.
2. P. D. Stein, A. Y. Yaekoub, F. Matta und M. Kleerekoper, «Fat embolism syndrome,» American journal of the medical sciences, 336(6), pp. 472-477, 2008.
3. E. M. Bulger, D. G. Smith, R. v. Maier und G. J. Jurkovich, «Fat embolism syndrom; a 10-Year review background,» The archives of surgery; 132 (4), pp. 435-439, 1997.
4. A. Pell, D. Hughes, J. Keating, J. Christie, A. Busuttil und G. Sutherland, «Brief report: fulminating fat embolism syndrome caused by paradoxical embolism through a patent foramen ovale,» New england journal of medicine; 329 (13), pp. 926-929, 1993.
5. C. A. Sulek, L. K. Davies, F. Kayser Enneking, P. A. Gearen und E. B. Lobato, «Cerebral microembolism diagnosed by transcranial doppler during total knee arthoplasty, correlation with transesophageal echocardiography,» Anesthesiology; 91 (3), pp. 672-676, 1999.
6. D. Sethi, S. Kajal und A. Saxena, «Neuroimaging findings in a case of cerebral fat embolism syndrome with delayed recovery,» Indian Journal of critical care medicine; 19(11), pp. 674-677, 2015.
7. R. Saigal, M. Mittal, A. Kansal, Y. Singh, P. R. Kolar und S. Jain, «Fat embolism syndrom,» Journal of the association of physicians of india, pp. 245-249, 2008.
8. C. Couturier, G. Dupont, F. Vassal, C. Boutet und J. Morel, «Effectiveness of decompressive hemicraniectomy to treat a life-threatening cerebral fat embolism,» Case reports in critical care, 2019.
9. S. A. Schonfeld, R. Ploysongsang und J. D. e. a. Dilisio, «Fat embolism prophylaxis with corticosteroids, a prospective study in high-risk patients,» Annals of internal medicine, pp. 438-443, 1983.
10. B. Lindeque, H. Schoeman, G. Dommisse, M. Boeyens und A. Vlok, «Fat embolism and the fat embolism syndrome. A double-blind therapeutic study.,» The journal of bone and joint surgery; 69(1), pp. 128-131, 1987 .
11. M. Kwiatt und M. Seamon, «Fat embolism syndrome,» International journal of critical illness and injury science; 3(1) , pp. 64-68, 2013.
12. P. Wöhler, B. Hirl und W. Kellermann, «Kasuistik – Zerebrales Fettemboliesyndrom nach beidseitiger Oberschenkelfraktur,» Anasthesiologie Intensivmedizin Notfallmedizin Schmerztherapie; 48(5), p. 300–302, 2013.
13. K. H. Kuo, Y. J. Pan, Y. J. Lai, W. K. Cheung, F. C. Chang und J. Jarosz, «Dynamic MR imaging patterns of cerebral fat embolism: A systematic review with illustrative cases,» American Journal of Neuroradiology, p. pp. 1052–1057, 2014.
14. O. Giyab, B. Balogh, P. Bogner, O. Gergely und A. Tóth, «Microbleeds show a characteristic distribution in cerebral fat embolism.,» Insights into Imaging, pp. 1-12, 2021.
15. S. Bederman, M. Bhandari, M. McKee und E. Schemitsch, «Do corticosteroids reduce the risk of fat embolism in patient with long bone fractures? A meta-analysis.,» Canadian journal of surgery; 52 (5), pp. 386-393 , 2009.
16. T. Kubota, T. Ebina, M. Tonosaki, H. Ishihara und A. Matsuki, «Rapid improvement of respiratory symptoms associated with fat embolism by high-dose methylpredonisolone: A case report,» Journal of Anesthesia; 17(3) , p. 186–189, 2003.