Wir beschreiben den Fall eines Patienten mit einem kleinzelligen Bronchuskarzinom, welcher seit sechs Jahren, jedoch verstärkt seit sechs Monaten zunehmend unter ausgeprägtem Schwindel mit Koordinationsstörungen litt. Nach diversen Abklärungen konnte als Ursache dafür ein paraneoplastisches neurologisches Syndrom (PNS) mit Hu-Antikörpern eruiert werden. PNS sind verschiedene neurologische Störungen, welche häufig in bestimmten Mustern auftreten. Die Ursache ist dabei immunvermittelt durch einen Tumor, weshalb die Therapie der PNS auch die Behandlung des zugrunde liegenden Tumors umfasst.
Paraneoplastische neurologische Syndrome (PNS) sind definiert als neurologische Störungen, welche jeden Teil des Nervensystems betreffen können und häufig in einem stereotypen Muster auftreten (1). Sie sind mit einem Tumor assoziiert und haben eine immunvermittelte Pathogenese. Es handelt sich um ein seltenes Krankheitsbild, das bei weniger als 1 % der Patienten mit einem Malignom beobachtet werden kann. Deshalb muss auch davon ausgegangen werden, dass die Diagnose regelmässig verpasst wird. Häufig treten die neurologischen Zeichen Monate bis Jahre vor der Diagnosestellung eines Tumors auf, werden jedoch zu diesem Zeitpunkt fehlgedeutet (2).
Anamnese
Der 65-jährige Patient leidet seit sechs Jahren an einer Schwindelsymptomatik mit Hörverlust des linken Ohres und einem Tinnitus auf der gleichen Seite. Diese Beschwerden wurden mehrfach als Morbus Menière bewertet.
In den letzten sechs Monaten wurde der Patient wiederholt wegen des Schwindels vorstellig. Neben der subjektiv klar verstärkten Schwindelsymptomatik kam es in den letzten Wochen zusätzlich zu einer begleitenden Gangunsicherheit. Es wurde ein MRI des Neurokraniums durchgeführt, welches regelrechte Strukturen darstellte. Durch die Kollegen der HNO wurde keine periphere Genese des Schwindels gefunden.
Ungefähr vier Monate nach Beginn des verstärkten Schwindels wurde im Rahmen einer ausgedehnten internistischen Abklärung die Diagnose eines metastasierten pulmonalen Tumorleidens mit lymphogenen und ossären Metastasen gestellt. Histologisch zeigte sich das Bild eines kleinzelligen Bronchuskarzinoms. Initial wurde eine tumorspezifische Kombinationstherapie mit Carboplatin, Etoposid und Durvalumab eingeleitet. Nach kurzer Therapiedauer kam es zu einer raschen Verschlechterung des Allgemeinzustandes, aufgrund dessen von einer weiteren tumorspezifischen Therapie abgesehen wurde.
Status
Die Zuweisung auf die Palliativstation erfolgte zur symptomorientierten Therapie aufgrund des zunehmenden Schwindels mit Nausea und Emesis. Klinisch zeigte sich bei Eintritt eine progrediente Gangstörung mit gleichzeitigen sensiblen Defiziten des linken Unterschenkels, eine ausgeprägte Ataxie der Extremitäten und eine Dysarthrie. Weiter bestand ein dysmetrischer Finger-Nase-Versuch links sowie ein Absinken des linken Armes im Armvorhalteversuch, ein feinschlägiger Tremor und eine verminderte Kraft des linken Beines mit Standunsicherheit und linksseitigen Ausfallschritten.
Verlauf
Wir leiteten eine stufenweise ausgebaute Therapie der Nausea mit Domperidon, Haloperidol, Cinnarizin, Betahistin und Dexamethason ein, welche jedoch keine zufriedenstellende Besserung der Symptomatik brachte. Durch die konsiliarisch hinzugezogenen Neurologen wurde die Diagnose einer sensibel betonten axonal-demyelinisierenden Polyneuropathie gestellt, wobei die axonale Komponente massgeblich mit der cisplatinhaltigen Chemotherapie assoziiert war. Die Gangstörung erschien diesbezüglich jedoch zu ausgeprägt.
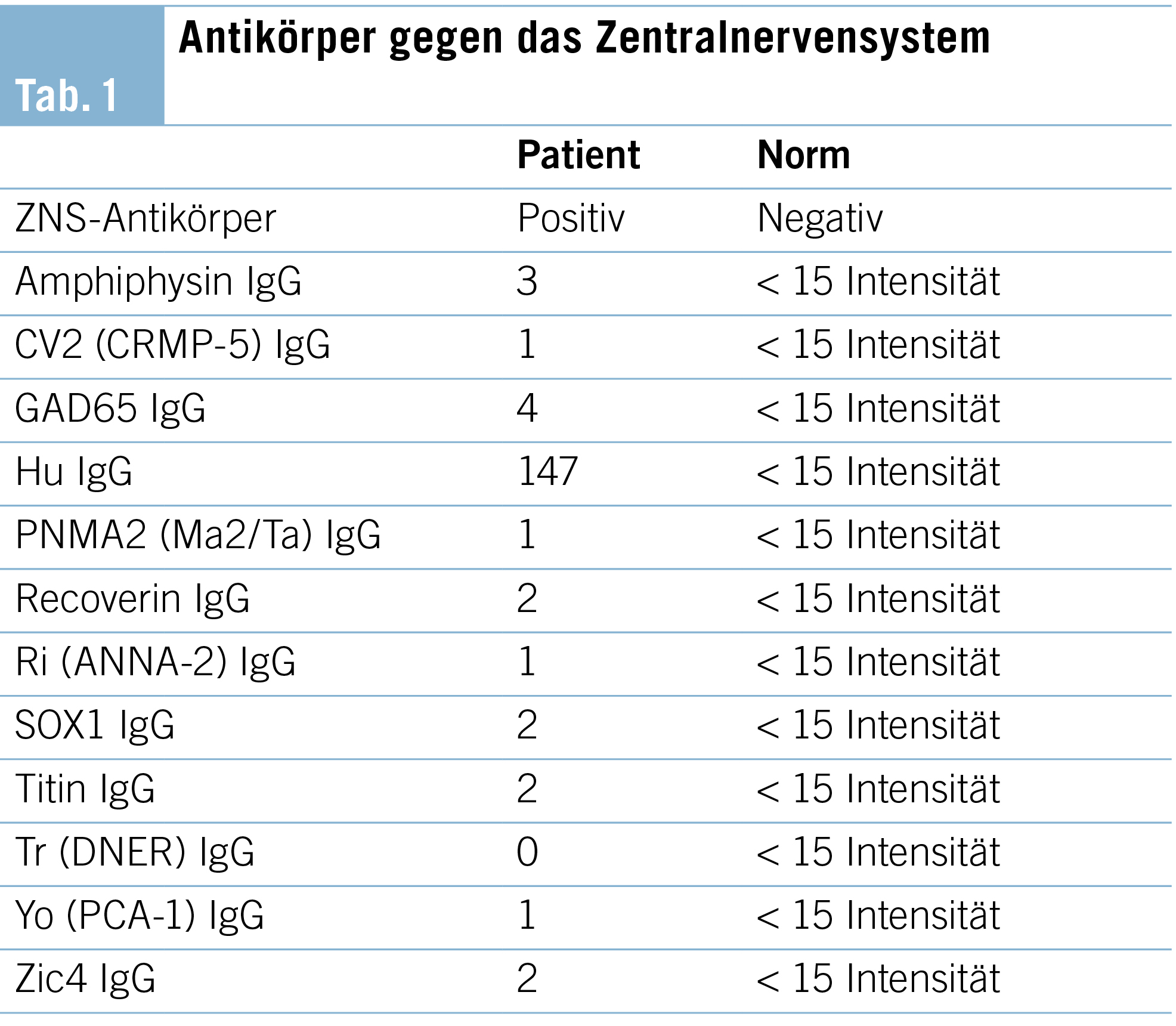
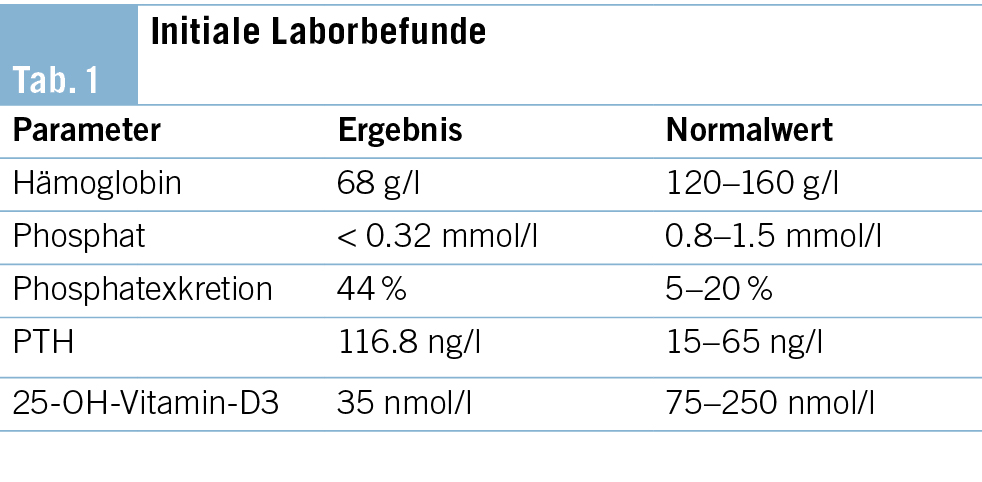
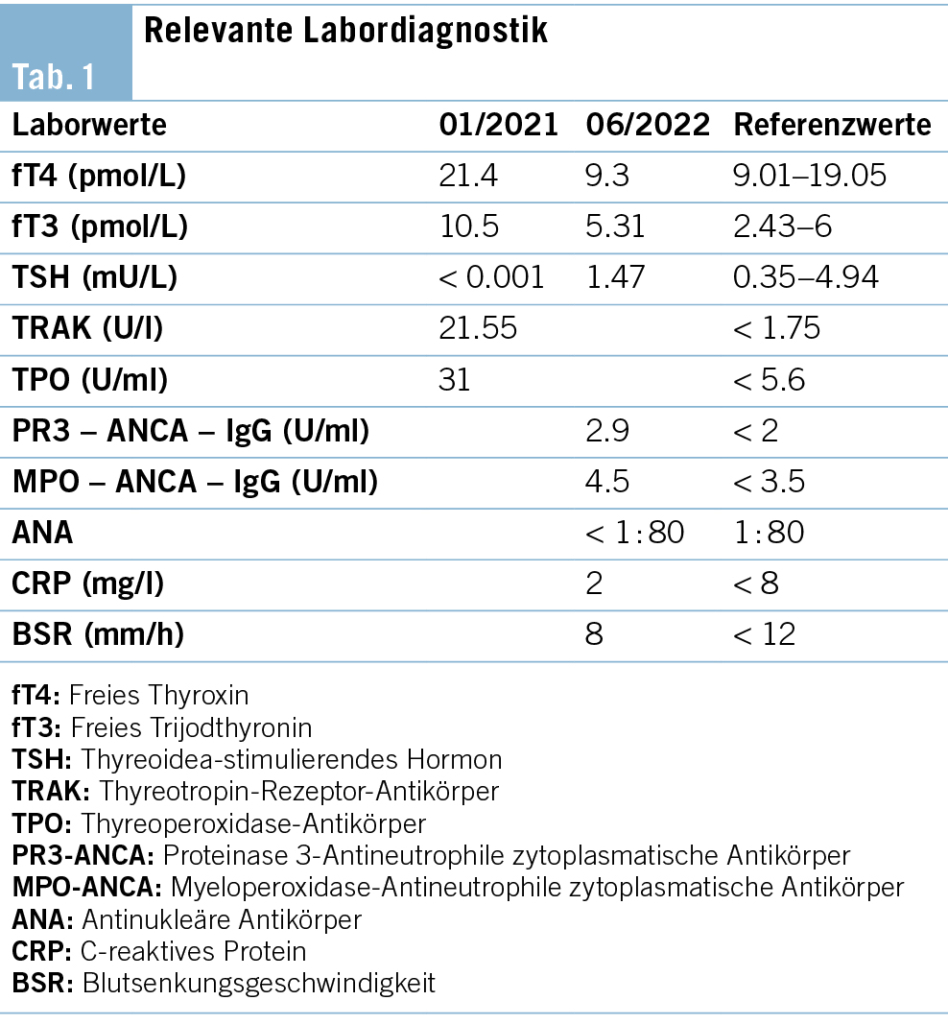
Die weiter gehenden Abklärungen zeigten positive Antikörper gegen das Zentralnervensystem, welche sich in der Differenzierung als Anti-Hu-Antikörper mit einer Intensität von 147 (Norm < 15) erwiesen. Sämtliche weiter bestimmte Antikörper waren negativ. Somit konnten wir die Diagnose eines paraneoplastischen Anti-Hu-Syndroms stellen, mit welchem die Beschwerden gut erklärt werden konnten, siehe Tabelle 1.
Therapie
Die Behandlung eines paraneoplastischen neurologischen Syndroms (PNS) liegt in der Behandlung des auslösenden Tumors. Eine immunsuppressive Therapie bringt, im Gegensatz zu anderen antikörpervermittelten Erkrankungen, jedoch keinen Benefit. In unserem Fall war wegen des stark reduzierten Zustandes keine kausale Therapie mehr möglich.
Bei zunehmender Symptomatik mit vollständiger Gangunfähigkeit, starker Dysarthrie und zunehmender Vergesslichkeit versuchten wir, ohne wesentlichen Erfolg, eine Steroidstosstherapie mit 1.g Methylprednisolon über drei Tage. Bei massiver Progredienz der Erkrankung war schlussendlich nur noch eine rein symptomatische Behandlung möglich, und es kam zu einem Versterben des Patienten drei Monate nach Diagnosestellung des kleinzelligen Bronchuskarzinoms.
Diskussion
Ein Expertengremium hat im Jahr 2021 eine neue Einteilung der paraneoplastischen neurologischen Syndrome (PNS) vorgeschlagen, wobei die Symptomkonstellationen in «Phänotypen mit intermediärem Risiko» und «Hochrisiko-Phänotypen» eingeteilt wurden (1). Das Risiko bezieht sich hierbei auf die Wahrscheinlichkeit, dass die Ätiologie der Symptome auf ein PNS zurückzuführen ist. Das Gremium hat dabei eine dreistufige Klassifizierung in definitiv, wahrscheinlich und möglich vorgenommen. Jede Stufe kann mithilfe des PNS-Care-Scores (Tab. 2) eingeschätzt werden. Dabei werden klinischer Phänotyp, Antikörpertyp, das Vorhandensein oder Nichtvorhandensein eines Malignoms und die Beobachtungszeit erfasst. Die eindeutige Diagnosestellung eines PNS erfordert das Vorhandensein von Antikörpern mit hohem oder mittlerem Risiko mit Ausnahme beim Opsoklonus-Myoklonus.
Zu den Hochrisiko-Phänotypen (früher: klassische PNS) gehören:
Die Enzephalomyelitis, die limbische Enzephalitis, das rapidly progressive zerebelläre Syndrom, der Opsoklonus-Myoklonus, die gastrointestinale Pseudoobstruktion (enterische Neuropathie), das Lambert-Eaton-Myasthenie-Syndrom sowie die sensorische Neuronopathie (1). Auf die einzelnen Krankheitsbilder gehen wir hier nicht ein.
Bei den «Phänotypen mit intermediärem Risiko» sollte vor allem an ein paraneoplastisches Geschehen gedacht werden, wenn keine plausiblen alternativen Diagnosen gefunden werden können und ein rascher Progress (weniger als drei Monate) sichtbar wird. Dies gilt auch dann, wenn Entzündungszeichen im Liquor oder MRI des Neurokraniums/Rückenmarks gefunden werden. Zu dieser Gruppe gehören beispielsweise eine Enzephalitis, welche nicht die Präsentation einer klassischen limbischen Enzephalitis aufweist, oder auch eine isolierte Myelopathie. Weiter gehört auch das Stiff-Person-Syndrom dazu, wobei es hier zu schmerzhaften Muskelspasmen kommt, welche spontan auftreten oder durch Bewegung oder äussere Reize getriggert werden können.
Für diese einzelnen paraneoplastischen neurologischen Syndrome können diverse Autoantikörper bestimmt werden. Hierbei unterscheidet man Antikörper mit hoher (> 70 %; früher onkoneurale Antikörper genannt), mittlerer (30–70 %), tiefer (< 30 %) oder fehlender Assoziation zu einem zugrunde liegenden Tumor.
Es ist wichtig, dass eine gezielte Testung vorgenommen wird, denn ein unüberlegtes Testen erhöht die Wahrscheinlichkeit für falsch positive und falsch negative Resultate. Serum und Liquor sollen auf Antikörper getestet werden, wobei insbesondere die IgG-Antikörper eine diagnostische Aussagekraft haben.
Anti-Hu- (ANNA-1)-Antikörper zeigen sich insbesondere beim Phänotyp einer sensorischen Neuropathie, einer Enzephalomyelitis oder auch einer gastrointestinalen Pseudoobstruktion. Die häufigsten Tumorarten sind das kleinzellige Lungenkarzinom und deutlich weniger häufig andere nicht kleinzellige Lungenkarzinome oder neuroendokrine Tumore (3, 4). Seltener können auch bei Patienten ohne ein Malignom Anti-Hu-Antikörper nachgewiesen werden (5).
In unserem Fall begannen die Beschwerden mit Schwindel, was in der Palliativmedizin ein häufiges Problem darstellt und oftmals schwierig zu behandeln und für die Betroffenen sehr beeinträchtigend ist. Vielfach kann die genaue Ätiologie nicht eruiert werden. Differenzialdiagnostisch vermuteten wir initial mögliche, bisher noch nicht sichtbare Hirnmetastasen und versuchten bei fehlenden kausalen Therapieoptionen, eine medikamentöse Behandlung des Schwindels einzuleiten. Eine zerebrale Bildgebung zeigte keine Hinweise für einen zerebralen Befall durch das bekannte kleinzellige Bronchuskarzinom. Im Rahmen der weiteren Abklärung konnten wir dann die Diagnose des Anti-Hu-Syndroms stellen, siehe Tabelle 2. Dies erbrachte mehr Klarheit und Verständnis für den Patienten und seine Angehörigen. Damit war jedoch auch klar, dass die Symptomatik und Koordinationsschwierigkeiten nur sehr schwer behandelbar sind.
Rückblickend lässt sich nicht genau klären, ob es sich bei der Schwindelsymptomatik vor sechs Jahren bereits um erste Anzeichen der Tumorerkrankung gehandelt hatte. Aufgrund der theoretischen Grundlagen der PNS wäre es möglich, jedoch war die klinische Präsentation unterschiedlich, weshalb es wahrscheinlich eine andere Ätiologie war. Eine so lange Latenzzeit von Erstsymptomatik bis zur Erstdiagnose erscheint zudem bei einem kleinzelligen Bronchuskarzinom sehr unwahrscheinlich.
Die Schwindelsymptomatik vier Monate vor klinischer Manifestation eines kleinzelligen Bronchuskarzinoms war, retrospektiv gesehen, mit hoher Wahrscheinlichkeit der Beginn des PNS. Ataxie, Stand- und Gangunsicherheit würden zu einer Kleinhirndegeneration passen, wobei das MRI, wie in unserem Fall, in einer Frühphase häufig normal ausfällt. Veränderungen in der Bildgebung, wie beispielsweise eine Kleinhirndegeneration, sind teilweise im späteren Verlauf zu sehen. Die Symptome können von Patienten und nicht spezialisierten Untersuchern als unspezifischer Schwindel gesehen werden und erlangen somit nicht die richtige Beachtung. Eine präzise Anamnese zur genauen Erfassung der Symptomatik ist von äusserster Wichtigkeit und kann bei der Diagnosestellung weiterhelfen.
Die diagnostizierte periphere Polyneuropathie, welche initial im Rahmen der cisplatinhaltigen Chemotherapie interpretiert wurde, könnte retrospektiv ebenfalls mit dem Anti-Hu-Syndrom assoziiert gewesen sein.
Für die Praxis
Paraneoplastische Syndrome sind selten und schwierig zu diagnostizieren.
Im Falle einer unklaren, länger dauernden Symptomatik des Schwindels sollen eine weiter gehende Abklärung und genaue Anamnese erfolgen.
Nach Ausschluss der üblichen Ursachen des Schwindels kann eine paraneoplastische Ursache mit Bestimmung der entsprechenden Antikörper evaluiert werden.
Die Behandlung richtet sich einerseits nach der Ursache, andererseits kann auch eine rein symptomatische Behandlung in Anbetracht der Tumorsituation gewählt werden.
Ein paraneoplastisches neurologisches Syndrom kann auch bei nicht bekanntem Malignom vorliegen.
Iris Neto
Palliative Care
Kantonsspital Graubünden
CH-7000 Chur
Dr. med. MSc Cristian Camartin
Leiter Palliative Care
Kantonsspital Graubünden
Loëstrasse 170 Chur
7000 Chur
cristian.camartin@ksgr.ch
Die Autorin und der Autor haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Graus F, Vogrig A, Muñiz-Castrillo S, Antoine JG, Desestret V et al. Updated Diagnostic Criteria for Paraneoplastic Neurologic Syndromes. Neurol Neuroimmunol Neuroinflamm. 2021 May 18;8(4):e1014.
2. Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003 Oct 16;349(16):1543-54.
3. Graus F. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124(pt 6):1138-1148.
4. Yu Z, Kryzer TJ, Griesmann GE, Kim K, Benarroch EE, Lennon VA. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol. 2001;49(2):146-154.
5. Honnorat J, Didelot A, Karantoni E, et al. Autoimmune limbic encephalopathy and anti-Hu antibodies in children without cancer. Neurology. 2013;80(24): 2226-2232.
Während Pankreasraumforderungen sonographisch schwierig zu detektieren sind, hilft die Schichtbildgebung enorm, fördert aber mit zunehmender Verfügbarkeit der Magnetresonanztomographie (MRI) zunehmend Inzidentalome zutage. Die Anamnese und Krankengeschichte ist nur bei zwei Entitäten charakteristisch. Eine bekannte akute/chronische Pankreatitis spricht für eine benigne Pseudozyste, eine vorhandene B-Symptomatik für einen malignen Tumor. Herausfordernder ist die Zuordnung asymptomatischer Zysten, welche ausgiebige Diagnostik und in fortgeschrittenen Stadien auch Pankreasresektionen nach sich ziehen können. Kommt es zur Überdiagnostik und – therapie, spricht man auch von VOMIT: victims of modern imaging technology (1). Auf der anderen Seite gilt es, diejenigen Präkanzerosen ohne Symptome zu identifizieren, welche einer Überwachung zugeführt werden sollen, mit dem Ziel, die Progression in Richtung eines invasiven Karzinoms rechtzeitig zu erkennen.www
Ziel dieser Übersicht ist, auf inzidentell nachgewiesene Pankreaszysten zu fokussieren mit Darstellung der Abklärungen sowie der weiteren Überwachung; nicht im Vordergrund stehen Pankreaskarzinom und Pseudozysten. Letztere werden im Sinne der Differenzialdiagnosen mit besprochen, da Pseudozysten im klinischen Alltag gelegentlich ebenfalls zufällig nachgewiesen werden, jedoch nicht überwacht werden sollten.
Bei einer 70-jährigen, bis auf eine arterielle Hypertonie sowie Übergewicht (BMI 27 kg/m2), gesunden Patientin wird aufgrund eines Fahrradunfalls eine Computertomographie (CT) durchgeführt, wo sich als Zufallsbefund eine 28 mm grosse zystische Pankreasläsion bei ansonsten normalem Pankreasparenchym nachweisen lässt. Die Patientin stellt sich in der Folge zur Besprechung des weiteren Vorgehens in der Sprechstunde vor.
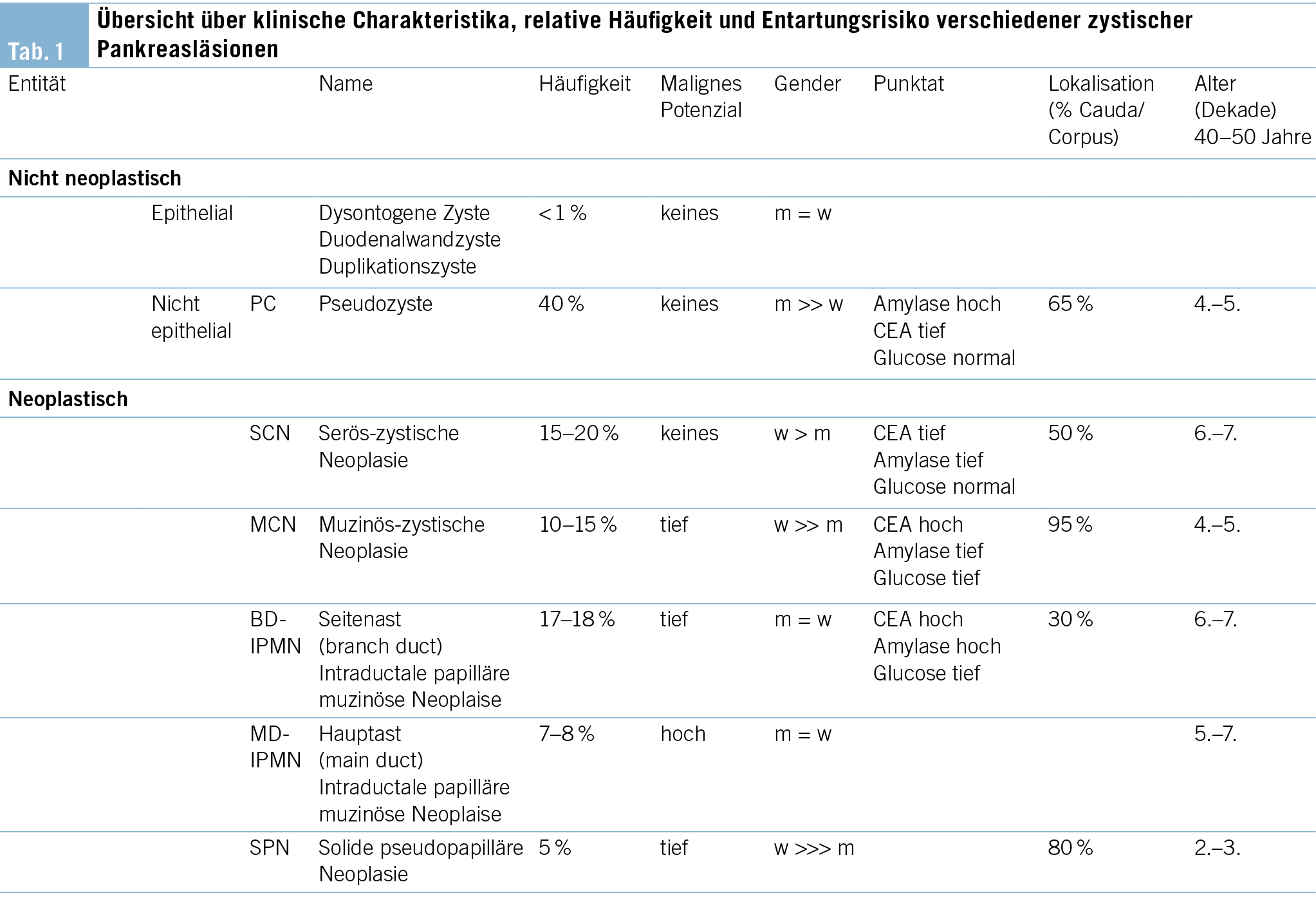
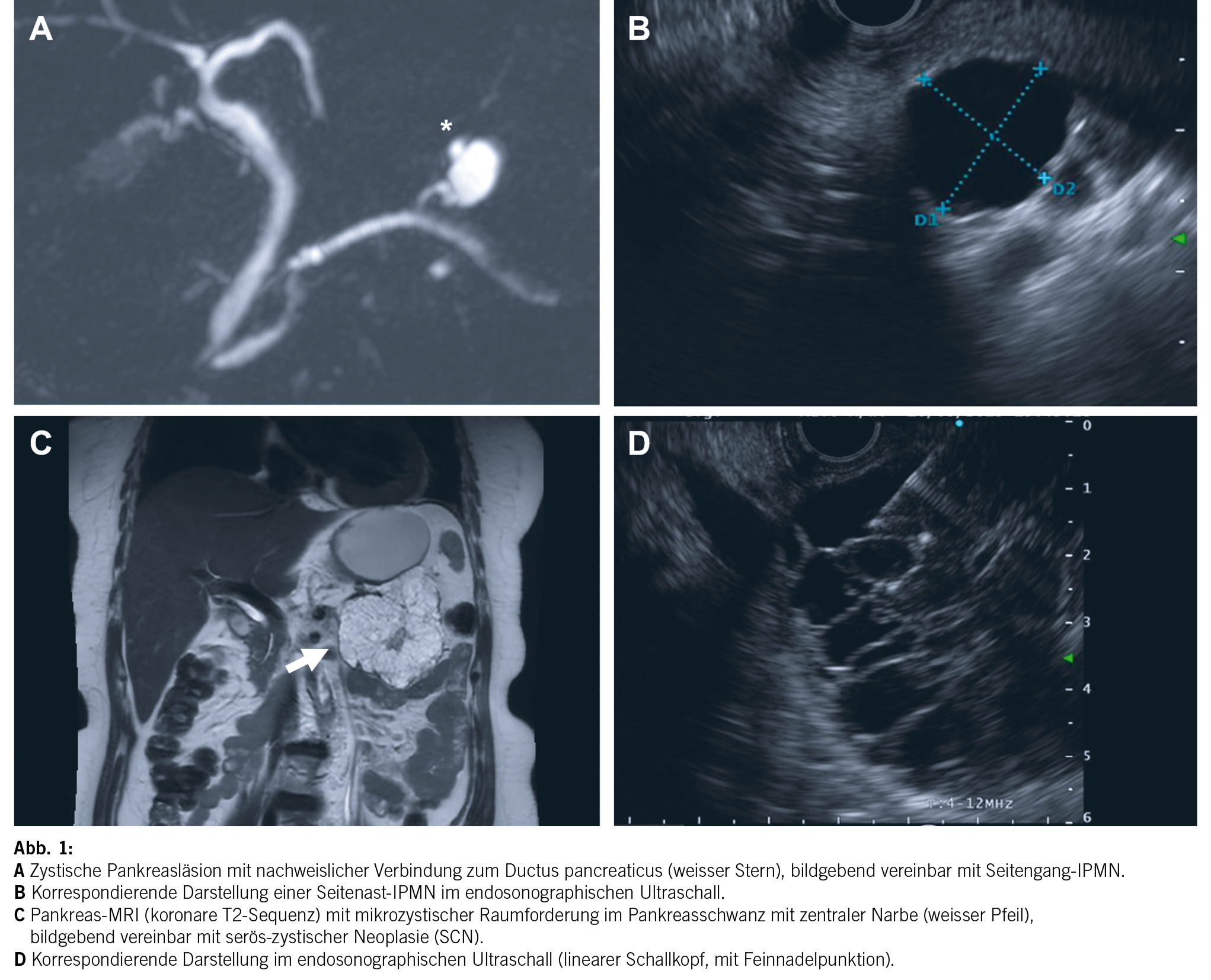
Aus klinischer Sicht stellt sich zunächst die Frage, wie hoch das maligne Potenzial der inzidentell nachgewiesenen zystischen Pankreasläsion zu beziffern ist. Während nicht neoplastische Entitäten wie Pseudozysten oder dysontogene Zysten keinerlei malignes Potenzial aufweisen, handelt es sich bei der Mehrheit der zufällig nachgewiesenen zystischen Pankreasläsionen um neoplastische Zysten respektive Präkanzerosen (Tab. 1). Dementsprechend steht in unserem Beispiel differenzialdiagnostisch eine neoplastische Zyste im Vordergrund, weshalb als nächster Schritt ein MRI durchgeführt wird (Abb. 1a/b), welches eine Verbindung der Läsion zum Gangsystem nachweist. Infolgedessen besteht der dringende Verdacht auf eine Seitenast-IPMN. Aufgrund der Grösse des Befundes entscheidet man sich ferner zur Durchführung einer oberen Endosonographie inkl. Kontrastmittelgabe, wo Noduli definitiv ausgeschlossen werden können (Abb. 1b). Die Analyse der endosonographisch gesteuerten punktierten Zystenflüssigkeit (hohes CEA, hohe Amylase, tiefe Glucose) lässt die Diagnose einer Seitenast-IPMN definitiv stellen. Aufgrund des erhöhten Risikos für die Entwicklung eines Pankreaskarzinoms wird die Aufnahme in ein Surveillance-Programm empfohlen.
Bei der Seitenast-IPMN handelt es sich mit ca. 80 % aller zufällig nachgewiesener zystischer Pankreasneoplasien um die häufigste Entität (2). Das maligne Potenzial hängt von verschiedenen Parametern ab. Wichtigste Kriterien diesbezüglich stellen die Grösse der Läsion, Dilatation des Hauptganges sowie Noduli dar. Hierauf basierend erfolgt eine Risikostratifikation, welche für die weitere Beratung der Patientinnen und Patienten essenziell ist. Diesbezüglich stellt die Arbeit von Mukewar et al. (3) die Verhältnisse sehr anschaulich dar. Während das Risiko eines Pankreaskarzinoms bei einer IPMN mit Hauptgangbeteiligung oder Nodulus als hoch zu bezeichnen ist (ca. 25 %/10 Jahre), ist das Risiko für Patienten mit Seitenast-IPMNs ohne weiterer radiologischer oder klinischer Risikofaktoren deutlich kleiner, relativ zur Normalbevölkerung jedoch immer noch deutlich erhöht (ca. 8 %/ 10 Jahre) (4), wobei das Risiko über die Jahre kontinuierlich ansteigt. Aufgrund der daraus abzuleitenden Indikation zur langfristigen Überwachung ist eine zweifelsfreie Diagnose essenziell. Insbesondere gutartige seröse Zystadenome (Abb. 1c/d) oder inzidentell nachgewiesene Pseudozysten müssen bildgeberisch oder ggf. auch mithilfe einer endosonographisch gesteuerten Punktion ausgeschlossen werden, damit unnötige, repetitive Verlaufskontrollen verhindert werden.
Diagnostik zystischer Pankreasläsionen: Stellenwert der Radiologie
Die primäre Aufgabe der Radiologie ist es, zystische Pankreasläsionen einerseits zu detektieren und andererseits wenn möglich zu charakterisieren. Wie oben erwähnt, werden zystische Pankreasläsionen häufig zufällig entdeckt. Der Nachweis von zystischen Pankreasläsionen in populations-basierten Studien hängt von der Art der Bildgebung ab und liegt bei der Kernspintomographie (MRI) bei bis zu 49 % der untersuchten Personen (5). Die Prävalenz der zystischen Läsionen und somit die Detektionsrate steigt mit dem Alter und liegt bei 61 % bei > 70 Jahren (6).
Wichtigste bildgebende Modalität zur Abklärung von zystischen Pankreasläsionen ist das MRI mittels umfassendem multiparametrischem Pankreasprotokoll. In diesem Zusammenhang hat sich die Magnetresonanz-Cholangiopankreatikographie (MRCP) als Standard etabliert, welche eine stark T2-gewichtete Sequenz verwendet. Strukturen mit hohem Wassergehalt stellen sich im Gegensatz zu den umgebenden Weichteilstrukturen hyperintens, d. h. hell, dar, während die umgebenden Weichteilstrukturen dunkel bleiben. Dies führt zum gewollten Aspekt der hellen Darstellung des Pankreasganges, der Gallengänge und auch der zystischen Pankreasläsionen auf dunklem Hintergrund (Abb. 1a).
Das MRI mit MRCP ist sowohl bei der Detektion als auch bei der Charakterisierung von zystischen Pankreasläsionen sowohl der Computertomographie (CT) als auch der transabdominalen Sonographie überlegen (7, 8). Auch die fehlende Applikation ionisierender Strahlung ist bei der Notwendigkeit von teilweise langjährigen Verlaufskontrollen ein wichtiger Faktor.
Radiologische Differenzialdiagnose
Im Folgenden sollen die typischen bildgebenden Eigenschaften der häufigsten zystischen Pankreasneoplasien (Tab. 1) beschrieben werden. Die bereits oben erwähnte intraduktale papilläre muzinöse Neoplasie (IPMN) kann sich in drei verschiedenen Formen manifestieren, als Seitengang-, als Hauptgang-IPMN oder als gemischte Form. Sie stellt sich als zystische (T2-hyperintense) Dilatation der besagten Gänge dar, d. h. bei Befall der Seitengänge als uni- oder mulitlokuläre, lobulierte, traubenartige Läsion, bei Befall des Hauptgangs in Form als segmentale oder diffuse Dilatation des Pankreashauptganges ≥ 5mm. Ein spezifisches bildgebendes Kriterium, das die IPMN von den anderen zystischen Neoplasien unterscheidet, ist die nachweisliche Verbindung zum Gangsystem (Abb. 1a). Eine typische Eigenschaft der Seitengang-IPMN ist ausserdem die Multiplizität.
Die muzinös-zystische Neoplasie (MCN) tritt fast ausschliesslich bei Frauen auf, mit höchster Inzidenz in der 5. Dekade. Klinisch ist das Erkennen dieser Entität wichtig, da diese Läsionen ein mit der Hauptast-IPMN vergleichbares Malignitätsrisiko aufweisen (9, 10). Die Lokalisation ist häufig im Pankreascorpus oder -cauda. Eine Kommunikation zum Ductus pancreaticus besteht, im Gegensatz zu den IPMN, nicht. Bildmorphologisch besteht meistens eine uni- oder multilokuläre Makrozyste (> 2cm) mit oder ohne Septierungen, teils mit «Zyste-in-Zyste»-Aspekt. Die äussere Begrenzung ist meistens rund, im Gegensatz zur eher lobulierten Seitengang-IPMN. In 25 % finden sich murale Verkalkungen.
Die serös-zystische Neoplasie (SCN) (Abb. 1c/d) kann sich in Form verschiedener Phänotypen manifestieren. Der typische Aspekt ist allerdings der mikrozystische (Zysten < 2 cm) oder Honigwaben-Aspekt. Die Zysten sind oft so klein, dass in der CT der Eindruck einer soliden Läsion entsteht. Ausserdem typisch ist eine zentrale, manchmal verkalkte Narbe, welche sich computertomographisch bestätigen lässt. Erschwerend kommen in ca. 10 % auch atypische, rein makrozystische Formen der SCN vor, welche bildgebend nicht von einer muzinösen Läsion mit Malignitätspotenzial unterschieden werden können.
Die solid-pseudopapilläre Neoplasie (SPN) ist eine seltene Läsion und kommt fast nur bei jungen Frauen in der 2.–3. Dekade vor. Es handelt sich hier um eine gemischt solid-zystische Neoplasie mit Nekrosen und hämorrhagischen Anteilen, typischerweise in der Cauda lokalisiert. Sie sind meist asymptomatisch und werden als Zufallsbefund in der Schnittbildgebung entdeckt.
Bei Pseudozysten handelt es sich typischerweise um hypodense zystische Läsionen mit flächigem Kontakt zum Pankreas. Die Wand der Pseudozyste ist in der Regel zart. Im MRI lässt sich im Gegensatz zur Seitengang-IPMN keine Verbindung zum Gangsystem nachweisen. Nebst der Anamnese mit St. n. Pankreatitis ist das Aspirat des Zysteninhalts charakteristisch, in dem die Amylase bei gleichzeitig tiefem CEA stark erhöht ist (Tab. 1).
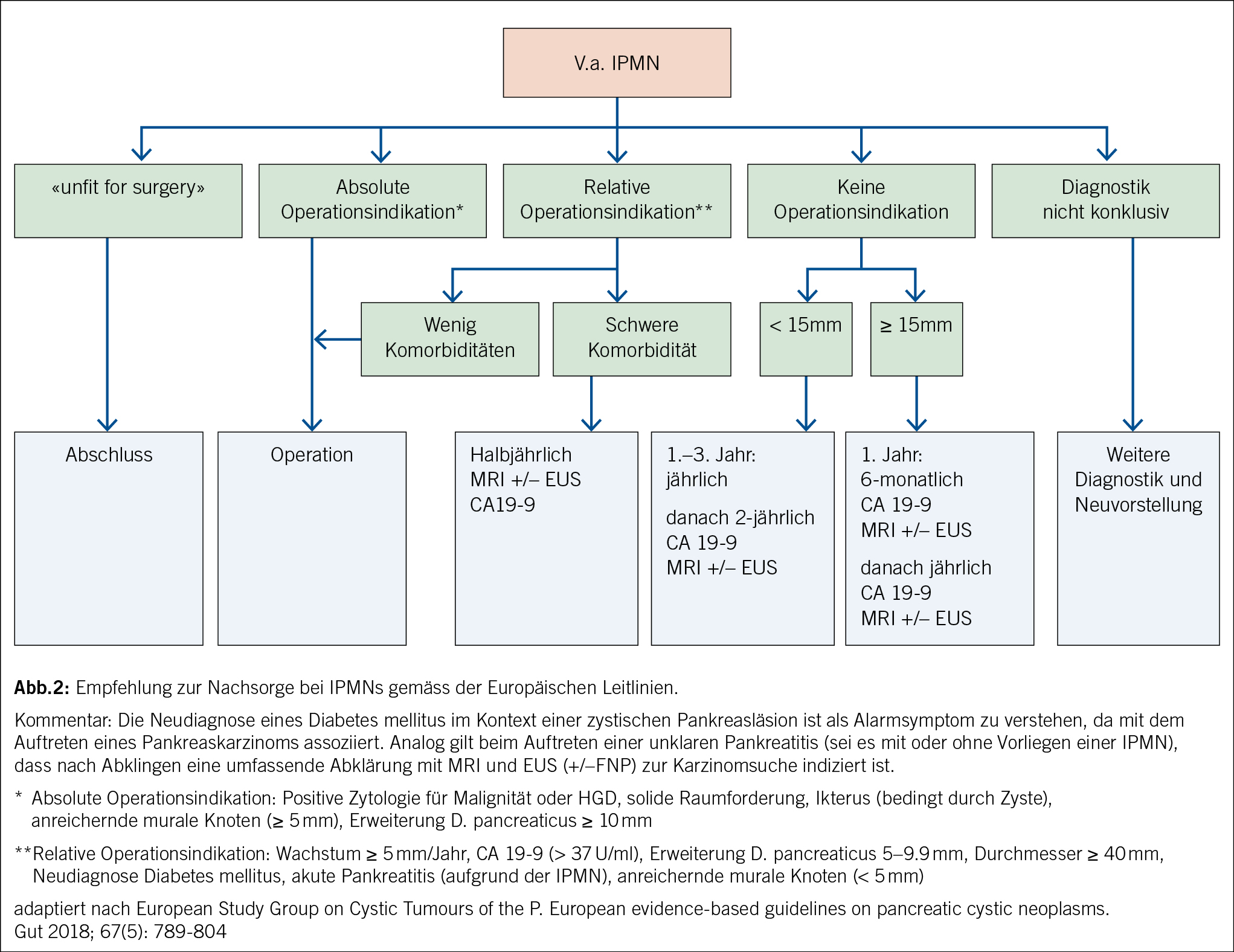
Nebst der Charakterisierung einer zystischen Pankreasläsion ist es auch Aufgabe der Radiologie, nach bildgebenden Zeichen zu suchen, welche auf ein erhöhtes Malignitätsrisiko weisen. Diese bildgebenden Zeichen werden in verschiedenen Leitlinien meist in einem zwei- oder mehrstufigen System abgebildet. In den European evidence-based guidelines on pancreatic cystic neoplasms (11) werden bei radiologisch vermuteter IPMN folgende bildgebende Zeichen als «relative Indikation für Chirurgie» bezeichnet (Abb. 2): Wachstumsrate der zystischen Läsion von ≥ 5 mm/Jahr, eine Dilatation des Pankreashauptganges von 5–9.9 mm, Zystendurchmesser von ≥ 4 cm und kontrastmittelaufnehmende murale Knoten von < 5 mm.
Rolle der gastroenterologischen Abklärungen mit Endoskopie/Endosonographie
Der Stellenwert der Endosonographie (EUS) besteht ergänzend zur MRI-Untersuchung und beantwortet bei Unklarheiten bezüglich Entität diese mithilfe mitunter einer Feinnadelpunktion.
Fokussiert zur Differenzierung der benignen Zysten SCA, MCA und IPMN kann die Punktion mit Bestimmung CEA, Amylase und Glucose nebst der Zytologie relevant sein. Hohe Amylasewerte im Punktat beweisen den Anschluss an den Pankreasgang bei der IPMN oder sprechen bei gleichzeitig normalen CEA-Werten für eine Pseudozyste. Hohe CEA-Werte bei normwertiger Amylase hingegen deuten auf ein MCA. In den letzten Jahren konnte gezeigt werden, dass eine tiefe Glucose (< 2.8 mmol/l) prädiktiv für das Vorliegen einer muzinösen Neoplasie ist (12). In naher Zukunft dürfte sich ferner die «Next Generation Sequencing» aus dem Aspirat als Standardmethode etablieren, mit welcher molekulargenetisch die Läsion zweifelsfrei charakterisiert werden kann.
Bei Knoten in Zysten können diese im Gegensatz zum MRI mit Kontrast-EUS von verklumptem Mukus unterschieden werden. Die EUS-gesteuerte Punktion erlaubt zudem gelegentlich den Nachweis dysplastischer oder malignen Zellen.
Die Indikation für eine EUS besteht somit bei Unklarheit betreffend Dignität, Entität oder aber bei Grössenwachstum und im Zusammenhang mit den oben beschriebenen Zeichen mit erhöhtem Malignitätsirisiko.
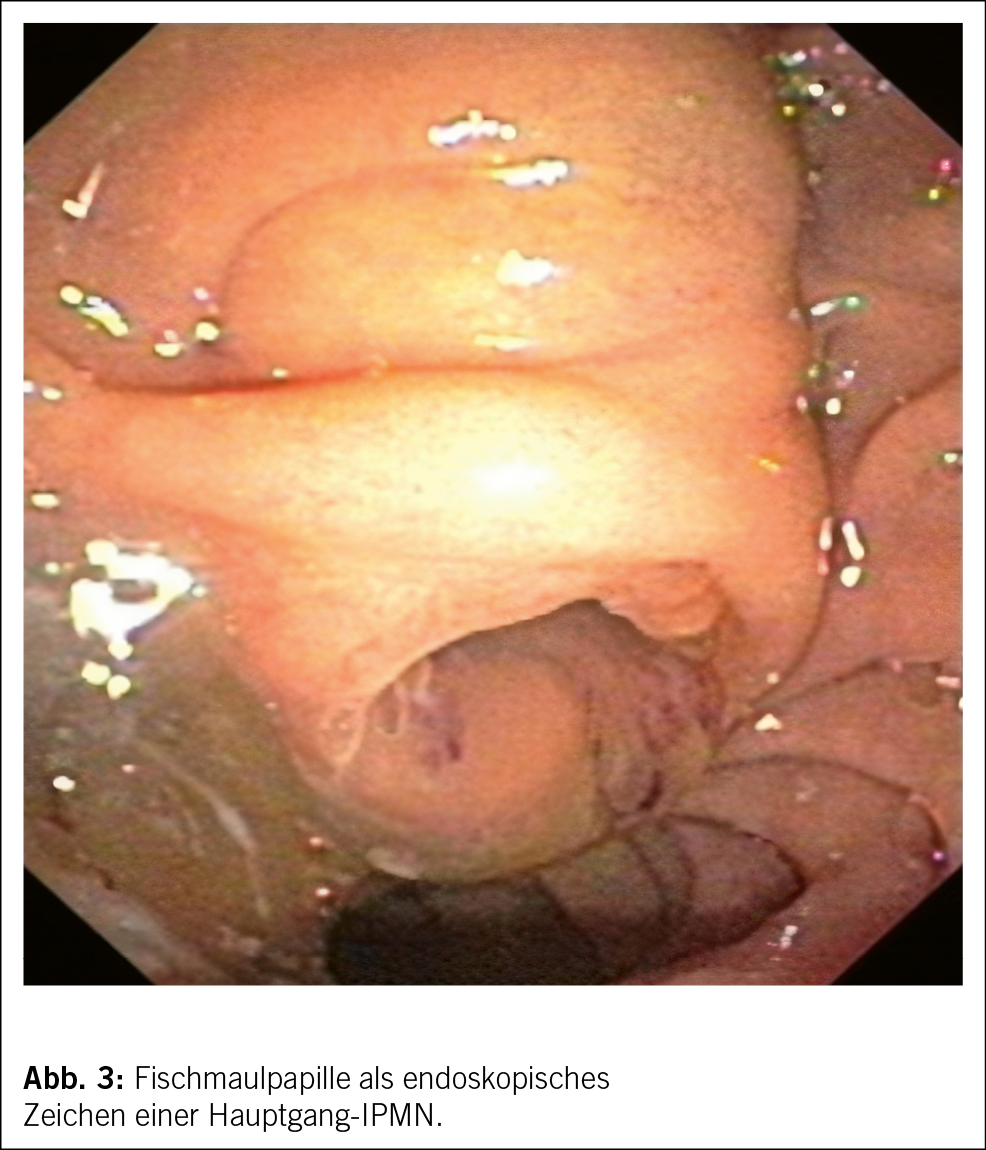
Die Duodenoskopie mit echter Seitblickoptik beurteilt die Papille. Eine sogenannte Fischmaulpapille (Abb. 3) ist pathognomonisch für eine Hauptast-IPMN und führt direkt zur Diskussion der chirurgischen Resektion.
Leitlinien und Überwachung
In den letzten Jahren wurden verschiedene Leitlinien zur Surveillance von Pankreaszysten, im speziellen IPMN, publiziert. Zuletzt wurde im Dezember 2023 eine weitere revidierte Version der «Fukuoka»-Leitlinien veröffentlicht (13), in welcher die Rolle der EUS noch etwas stärker gewichtet und ein adaptierter Abklärungsalgorithmus vorgeschlagen wird. Im Zentrum dieser Publikationen stehen radiologische Merkmale, welche auf ein klinisch signifikantes Malignitätsrisiko weisen. Da Pankreasresektionen im Allgemeinen mit einer verhältnismässig hohen Morbidität vergesellschaftet sind, sollten die Leitlinien gleichzeitig unnötige Resektionen verhindern. Anhand von klinischen Zeichen (u. a. Ikterus, Pankreatitis), radiologischen Befunden (z. B. Zystengrösse, Weite D. pancreaticus, Noduli), Tumormarker (CA19-9) und in selektiven Fällen zytologischen oder histologischen Untersuchungen wird versucht, eine Risikostratifikation vorzunehmen.
Dabei bestehen insbesondere bezüglich der Indikationsstellung zur Resektion resp. Durchführung der Surveillance bezüglich Modalität, dem Intervall und der absoluten Dauer der Nachsorge deutliche Unterschiede.
Die meistverwendeten Guidelines sind die
Revised International Consensus Guidelines (Fukuoka/Kyoto-Guidelines) (13, 14)
European evidence-based guidelines on pancreatic cystic neoplasms (11)
American Gastroenterology Association Guidelines (15)
Die Guidelines haben gemeinsam, dass sie zur Entdeckung High-grade-Dysplasien oder invasiver Karzinome eine relativ hohe Sensitivität besitzen, leider aber eine geringe Spezifität aufweisen. Dies führt tendenziell zu einer Überbehandlung der Patienten (17). Ein weiteres Problem in der Behandlung der Pankreaszysten ist die ungenügende Adhärenz zu den Guidelines. Daten aus der Literatur zeigen, dass nur ca. 2/3 aller der Patienten mit bekannten Pankreaszysten einen adäquaten Follow-up erhalten (18). Einen sehr lesenswerten Überblick über die verschiedenen Leitlinien resp. deren Unterschiede bietet die Arbeit von Aziz et al. (19).
Um ein strukturiertes Follow-up zu gewährleisten, aber auch da es sich um potenzielle Krebserkrankungen handelt, sehen wir eine interdisziplinäre Diskussion im Rahmen eines «Pankreaszystenboard» als empfehlenswert. Diese Diskussion soll einerseits unnötige Resektionen verhindern und andererseits eine adäquate und verhältnismässige Surveillance sicherstellen.
Ausblick
Die Häufigkeit zystischer Pankreasläsionen gerade bei älteren Patienten stellt eine klinische Herausforderung dar. Dem Risiko einer meist tödlich verlaufenden Karzinomerkrankung steht die Gefahr der «Überdiagnostik» mit damit assoziierter psychischer Belastung Betroffener und unnötigen Pankreasresektionen gegenüber. Dazuzurechnen sind Belastungen für das Gesundheitswesen finanzieller und logistischer Art in einem Gesamtsystem, in welchem in Zukunft die Ressourcen knapp werden dürften.
Die interdisziplinäre Diskussion der radiologischen resp. endosonographischen Befunde unter Berücksichtigung der Europäischen Leitlinien bietet Rahmenbedingungen, welche eine angemessene Überwachung oder chirurgische Therapie ermöglichen.
Unter welchen Umständen diese Überwachung bei einem gesunden Patienten beendet werden darf, ist ebenfalls noch umstritten. Erste Studien empfehlen jedoch, bei einem stabilen Verlauf über 5 Jahre und einer maximalen Zystengrösse von 30 mm bei über 75-Jährigen bzw. 15 mm bei über 65-Jährigen die Überwachung einzustellen (20).
Key Messages
• Symptomatische Pankreaszysten sind oft Pseudozysten.
• Asymptomatische Zysten sind in der Regel echte Neoplasien, am häufigsten IPMN vom Seitenast-Typ (BD-IPMN), welche zumeist ein niedriges Entartungsrisiko aufweisen.
• Eine Hauptast-IPMN (MD-IPMN) > 10 mm, anreichernde murale Knoten (≥ 5 mm) oder eine positive Zytologie stellen eine Indikation zur chirurgischen Resektion dar.
• Vor Abklärung oder Surveillance muss geklärt sein, ob eine chirurgische Therapie infrage käme. Im negativen Fall soll der Fall abgeschlossen werden.
• Die Nachsorge wird mittels MRCP seriell über Jahre vollzogen, wobei sich die Intervalle anhand der Risikostratifikation bemessen.
• Die obere Endosonographie hilft beim Nachweis von Zystencharakteristika, welche für eine Operation sprechen.
• Bei absoluter und insbesondere auch bei relativer Operationsindikation sollte der Patient an ein HSM-Zentrum zur weiteren Evaluation zugewiesen werden.
Dr. med. Bernhard Morell
Abteilung für Gastroenterologie und Hepatologie
Stadtspital Zürich
Birmensdorferstrasse 497
8063 Zürich
bernhardkaspar.morell@stadtspital.ch
Dr. med. Stephan Ullrich
Leitender Arzt
Institut für Radiologie und Nuklearmedizin
Birmensdorferstrasse 497
8063 Zürich
Dr. med. Stefan Gutknecht
Leitender Arzt
Klinik für Viszeral-, Thorax- und Gefässchirurgie
Stadtspital Zürich Triemli
Birmensdorferstrasse 497
8063 Zürich
Dr. med. Pascal Weibel
Facharzt für Chirurgie
Klinik für Viszeral- Thorax- und Gefässchirurgie
Stadtspital Zürich
Dr. med. Jörg Wydler
Leitender Arzt, Klinik Viszeral-, Thorax- und Gefässchirurgie
Chefarzt Klinik für Belegärzte
Stadtspital Zürich Triemli
Birmensdorferstrasse 497
8063 Zürich
Die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
• Symptomatische Pankreaszysten sind oft Pseudozysten.
• Asymptomatische Zysten sind in der Regel echte Neoplasien, am häufigsten IPMN vom Seitenast-Typ (BD-IPMN), welche zumeist ein niedriges Entartungsrisiko aufweisen.
• Eine Hauptast-IPMN (MD-IPMN) > 10 mm, anreichernde murale Knoten (≥ 5 mm) oder eine positive Zytologie stellen eine Indikation zur chirurgischen Resektion dar.
• Vor Abklärung oder Surveillance muss geklärt sein, ob eine chirurgische Therapie infrage käme. Im negativen Fall soll der Fall abgeschlossen werden.
• Die Nachsorge wird mittels MRCP seriell über Jahre vollzogen, wobei sich die Intervalle anhand der Risikostratifikation bemessen.
• Die obere Endosonographie hilft beim Nachweis von Zystencharakteristika, welche für eine Operation sprechen.
• Bei absoluter und insbesondere auch bei relativer Operationsindikation sollte der Patient an ein HSM-Zentrum zur weiteren Evaluation zugewiesen werden.
1. Shaikh U, Lewis-Jones H: Commercial CT scans: VOMIT victim of medical investigative technology. BMJ 2008, 336(7634):8.
2. Chang YR, Park JK, Jang JY, Kwon W, Yoon JH, Kim SW: Incidental pancreatic cystic neoplasms in an asymptomatic healthy population of 21,745 individuals: Large-scale, single-center cohort study. Medicine (Baltimore) 2016, 95(51):e5535.
3. Mukewar S, de Pretis N, Aryal-Khanal A, Ahmed N, Sah R, Enders F, Larson JJ, Levy MJ, Takahashi N, Topazian M et al: Fukuoka criteria accurately predict risk for adverse outcomes during follow-up of pancreatic cysts presumed to be intraductal papillary mucinous neoplasms. Gut 2017, 66(10):1811-1817.
4. Choi SH, Park SH, Kim KW, Lee JY, Lee SS: Progression of Unresected Intraductal Papillary Mucinous Neoplasms of the Pancreas to Cancer: A Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol 2017, 15(10):1509-1520 e1504.
5. Kromrey ML, Bulow R, Hubner J, Paperlein C, Lerch MM, Ittermann T, Volzke H, Mayerle J, Kuhn JP: Prospective study on the incidence, prevalence and 5-year pancreatic-related mortality of pancreatic cysts in a population-based study. Gut 2018, 67(1):138-145.
6. Moris M, Bridges MD, Pooley RA, Raimondo M, Woodward TA, Stauffer JA, Asbun HJ, Wallace MB: Association Between Advances in High-Resolution Cross-Section Imaging Technologies and Increase in Prevalence of Pancreatic Cysts From 2005 to 2014. Clin Gastroenterol Hepatol 2016, 14(4):585-593 e583.
7. Waters JA, Schmidt CM, Pinchot JW, White PB, Cummings OW, Pitt HA, Sandrasegaran K, Akisik F, Howard TJ, Nakeeb A et al: CT vs MRCP: optimal classification of IPMN type and extent. J Gastrointest Surg 2008, 12(1):101-109.
8. Sainani NI, Saokar A, Deshpande V, Fernandez-del Castillo C, Hahn P, Sahani DV: Comparative performance of MDCT and MRI with MR cholangiopancreatography in characterizing small pancreatic cysts. AJR Am J Roentgenol 2009, 193(3):722-731.
9. Zamboni G, Scarpa A, Bogina G, Iacono C, Bassi C, Talamini G, Sessa F, Capella C, Solcia E, Rickaert F et al: Mucinous cystic tumors of the pancreas: clinicopathological features, prognosis, and relationship to other mucinous cystic tumors. Am J Surg Pathol 1999, 23(4):410-422.
10. Reddy RP, Smyrk TC, Zapiach M, Levy MJ, Pearson RK, Clain JE, Farnell MB, Sarr MG, Chari ST: Pancreatic mucinous cystic neoplasm defined by ovarian stroma: demographics, clinical features, and prevalence of cancer. Clin Gastroenterol Hepatol 2004, 2(11):1026-1031.
11. European Study Group on Cystic Tumours of the P: European evidence-based guidelines on pancreatic cystic neoplasms. Gut 2018, 67(5):789-804.
12. McCarty TR, Garg R, Rustagi T: Pancreatic cyst fluid glucose in differentiating mucinous from nonmucinous pancreatic cysts: a systematic review and meta-analysis. Gastrointest Endosc 2021, 94(4):698-712 e696.
13. Tanaka M, Fernandez-Del Castillo C, Kamisawa T, Jang JY, Levy P, Ohtsuka T, Salvia R, Shimizu Y, Tada M, Wolfgang CL: Revisions of international consensus Fukuoka guidelines for the management of IPMN of the pancreas. Pancreatology 2017, 17(5):738-753.
14. Ohtsuka T, Fernandez-Del Castillo C, Furukawa T, Hijioka S, Jang JY, Lennon AM, Miyasaka Y, Ohno E, Salvia R, Wolfgang CL, Wood LD: International evidence-based Kyoto guidelines for the management of intraductal papillary mucinous neoplasm of the pancreas. Pancreatology 2024, 24(2):255-270.
15. Scheiman JM, Hwang JH, Moayyedi P: American gastroenterological association technical review on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015, 148(4):824-848 e822.
16. Hasan A, Visrodia K, Farrell JJ, Gonda TA: Overview and comparison of guidelines for management of pancreatic cystic neoplasms. World J Gastroenterol 2019, 25(31):4405-4413.
17. Canakis A, Maoz A, Tkacz JN, Huang C: Factors affecting the rates of adherence to surveillance recommendations for incidental pancreatic cystic lesions in a large urban safety net hospital. BMJ Open Gastroenterol 2020, 7(1).
18. Aziz H, Acher AW, Krishna SG, Cloyd JM, Pawlik TM: Comparison of Society Guidelines for the Management and Surveillance of Pancreatic Cysts: A Review. JAMA Surg 2022, 157(8):723-730.
19. Marchegiani G, Pollini T, Burelli A, Han Y, Jung HS, Kwon W, Rocha Castellanos DM, Crippa S, Belfiori G, Arcidiacono PG et al: Surveillance for Presumed BD-IPMN of the Pancreas: Stability, Size, and Age Identify Targets for Discontinuation. Gastroenterology 2023, 165(4):1016-1024 e1015.
Wir berichten über einen 89-jährigen Patienten, der sich aufgrund von Meläna und Leistungsminderung auf der Notfallstation vorstellte. Ursächlich für die Symptomatik war eine obere gastrointestinale Blutung aus einer Metastase eines follikulären Schilddrüsenkarzinoms, welches im Initialstadium im Jahr 1996 bereits operativ mittels Thyreoidektomie angegangen wurde.
Es erfolgte die abendliche notfallmässige Vorstellung eines 89-jährigen Patienten auf unserer Notfallstation aufgrund von Meläna in den letzten Tagen und einer begleitenden leichten Leistungsminderung. Der Patient war in einem guten Allgemeinzustand, wirkte biologisch deutlich jünger, und seine fixe Medikation bestand lediglich aus Eltroxin und Metoprolol. Gemäss seiner Aussage hatte er vor ca. 30 Jahren eine Schilddrüsenentfernung wegen eines Tumors sowie eine radikale Prostatektomie wegen eines Prostatakarzinoms vor 15 Jahren und einen Bluthochdruck. In der klinischen Untersuchung fanden sich bei leicht hypertensiven Blutdruckwerten keine anderweitigen pathologischen Befunde. Insbesondere die digital rektale Untersuchung war unauffällig und zeigte kein Blut am Fingerling. Im Labor fand sich eine hyporegenerative, normochrome, normozytäre Anämie mit einem Hämoglobin von 109 g/l (Normwert 140 bis 180 g/l). Es waren keine unmittelbaren Vorwerte vorhanden, sodass die Dynamik einer allfälligen Blutung offenblieb. Differenzialdiagnostisch schien ein eher chronischer gastrointestinaler Blutverlust mit Meläna vorzuliegen. Der Patient wurde stationär aufgenommen und eine endoskopische Untersuchung des oberen Gastrointestinaltraktes für den Folgemorgen vereinbart.
Abklärungsschritte
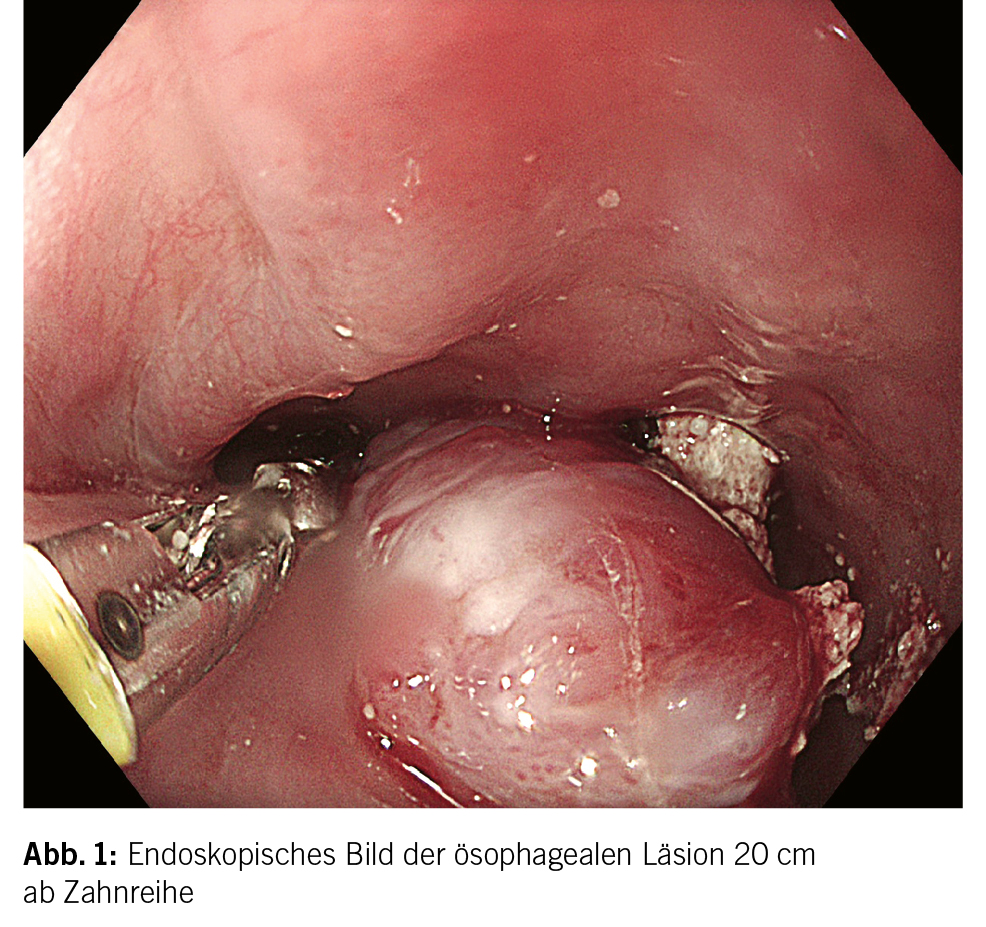
In der Gastroskopie am Folgetag fand sich, bei stabilem Hämoglobin über Nacht, der endoskopisch in Abb. 1 ersichtliche Befund im proximalen Ösophagus, wobei die restliche Untersuchung des oberen Gastrointestinaltraktes bis auf wenig Hämatinspuren im Magen unauffällig war.
Es handelte sich um einen das Lumen einengenden Befund im proximalen Ösophagus ungefähr 20 cm ab Zahnreihe mit makroskopisch scheinbar äusserst guter Durchblutung. Der Befund wurde aufgrund von diffusen Sickerblutungen bei kleinster Berührung mit dem Endoskop und der Biopsiezange vorerst nur mittels einem NaCl 0.9 % und 1:10 000 verdünnten Adrenalingemisch umspritzt. Die Endoskopie wurde beendet und der Patient für eine Computertomografie (CT)-Untersuchung des Halses, Thorax und Abdomens angemeldet, mit Frage nach Durchblutung und Ätiologie der Läsion.
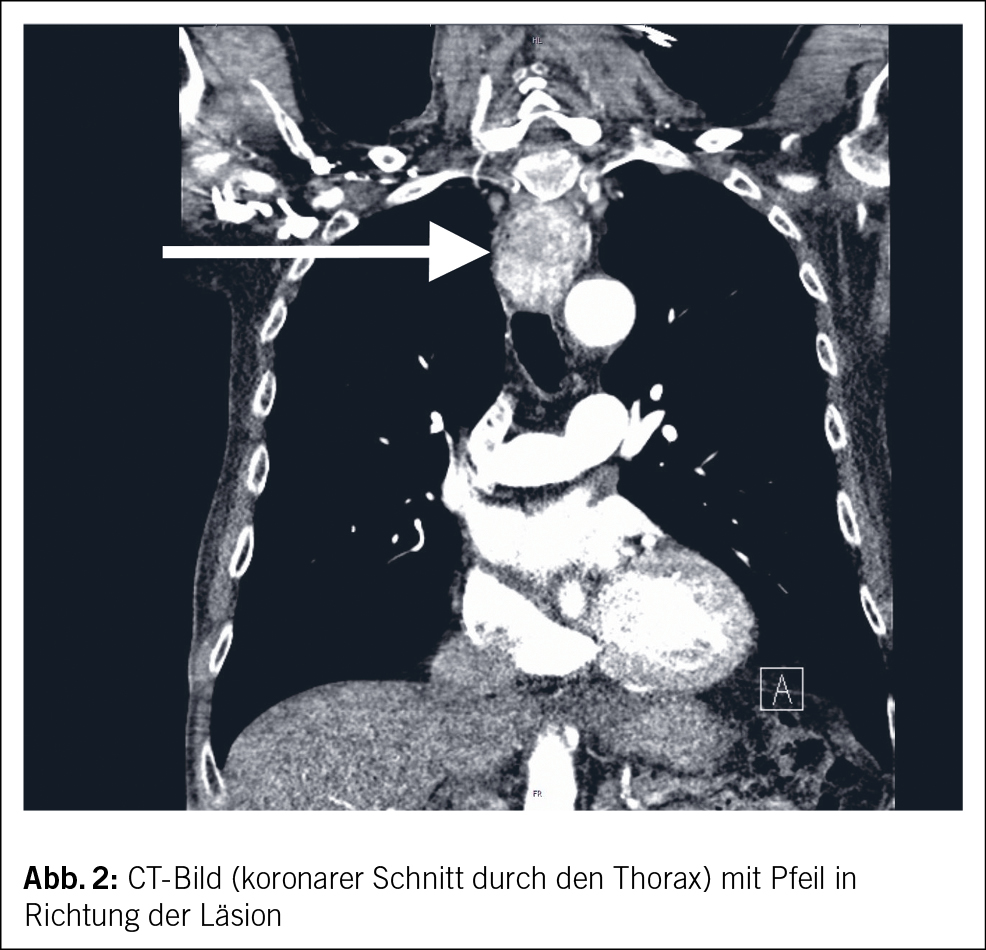
CT-grafisch wurde die Raumforderung dann als ungefähr 3 x 3 x 5 cm grosse Läsion im Bereich des proximalen Ösophagus mit deutlicher Kontrastmittelaufnahme beschrieben (Abb. 2). Die Ätiologie der Raumforderung blieb vorerst aber unklar. Differenzialdiagnostisch wurde von den Kollegen der Radiologie ein neuroendokriner Tumor, ein gastrointestinaler Stromatumor, ein Leiomyom oder ein ektopes, hyperproliferiertes Schilddrüsengewebe postuliert. Der Patient wurde auf der Station weiter klinisch überwacht, mittels hochdosierter Protonenpumpeninhibitoren (PPI) behandelt, und das Hämoglobin wurde engmaschig kontrolliert und eine erneute endoskopische Abklärung in Endosonografie-Bereitschaft geplant. In der erneuten gastroskopischen Untersuchung konnte der Befund nochmalig dargestellt werden, und es gelang die Entnahme einiger Proben zur histologischen Untersuchung. Auf eine obere Endosonografie wurde bei deutlicher Lumeneinengung durch den Befund vorerst verzichtet.
Diagnose
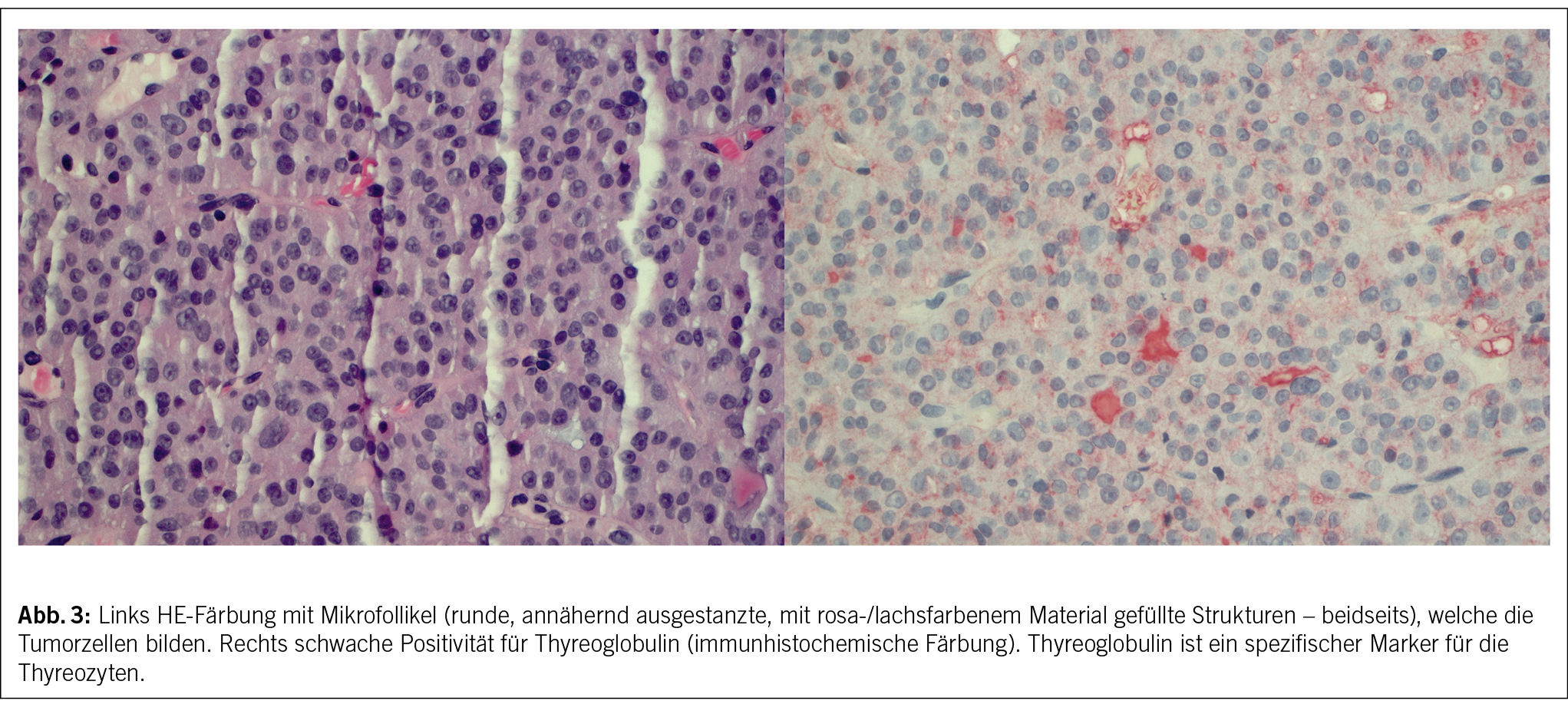
In der histologischen Aufarbeitung fanden sich Zellen eines Adenokarzinoms, morphologisch und immunhistochemisch vereinbar mit Metastasen eines follikulären Schilddrüsenkarzinoms (Abb. 3 links).
Verlauf
In der inzwischen natürlich erfolgten Durchsicht der Akten wurde der Patient im Jahr 1996, also vor fast 30 Jahren, aufgrund eines gut differenzierten follikulären Schilddrüsenkarzinoms komplett thyreoidektomiert.
Bis ins Jahr 2007 erfolgten insgesamt 3 Radiojodtherapien bei jeweils leichtem Anstieg des Thyreoglobulins. Posttherapeutisch erfolgte zuletzt im Jahr 2007 eine Jod-131-Ganzkörperszintigrafie ohne Mehranreicherung bei bereits damals leicht erhöhtem Thyreoglobulin (2.5 µg/l [Norm 1.6 bis 61.3µg/l]). Eine kleine Mehranreicherung paraösophageal links wurde im PET-CT im Jahr 2008 erstmalig beschrieben, und die Läsion in diesem Bereich war dann in einem PET-CT in 2015 grössenprogredient mit damals 1 x 1 cm. Endoskopisch und endosonografisch wurde diese Läsion damals abgeklärt und als mögliches Gefässkonvolut interpretiert. Weitere Abklärungen und Kontrollen diesbezüglich erfolgten nicht.
Der nun deutliche Anstieg des Thyreoglobulins auf 2782 µg/l (Norm 1.6 bis 61.3µg/l) mit zuletzt vorhandenem Wert von 9.5 µg/l im April 2008 und die immunhistochemisch bestätigte Expression von Thyreoglobulin in den Tumorzellen (Abb. 3 rechts) bestätigte nun eine Metastase des Schilddrüsenkarzinoms. Aktuell war die Läsion deutlich grösser (3.2 x 3.3 x 5.3 cm) als noch im Jahr 2015 (1 x 1 cm).
Die ösophageale Metastase bestand somit vermutlich seit mindestens 15 Jahren, war nun deutlich grössenprogredient und hatte aktuell zu einer Sickerblutung im Bereich der Läsion geführt. Eine Radiojodtherapie zum jetzigen Zeitpunkt war aufgrund der Grösse der Metastase, der bereits dreimal durchgeführten Radiojodtherapie und der fehlenden Jodaufnahme in der letzten posttherapeutischen Jod-131-Ganzkörperszintigrafie 2007 bei bereits damals erhöhtem Thyreoglobulin und nachgewiesener Pathologie im Bereich des Ösophagus nicht mehr möglich.
Aufgrund des Alters des Patienten sowie der Lokalisation der Metastase kam auch eine chirurgische Sanierung eher nicht infrage. Wir empfahlen dem Patienten darum eine Radiotherapie, mit welcher auch der Patient einverstanden war. Inzwischen wurde der Patient auch erfolgreich mit 25 x 2 Gy palliativ bestrahlt. Komplikationen des lokalen Befundes sind keine mehr aufgetreten. Die Bestrahlung wurde gut toleriert bis auf eine passagere Odynophagie.
Kommentar
Die Wichtigkeit der Anamnese und die Informationen betreffend der Vordiagnosen sind auch in diesem Fall eindrücklich. Die Anamnese mit Verdacht auf einen gastrointestinalen Blutverlust liessen uns rasch in die Richtung der Endoskopie als diagnostisches Mittel der Wahl denken. Endoskopisch fand sich dann eine Raumforderung im proximalen Ösophagus als Ursache. Die Diagnose des metastasierten Schilddrüsenkarzinoms liess sich histologisch, immunhistochemisch und bildgebend sichern.
In der Literatur finden sich sehr wenige Fallberichte mit ösophageal metastasierten follikulären Schilddrüsenkarzinomen. Lee et al. (1) berichteten über den zum damaligen Zeitpunkt (2008) offenbar erstmalig beschriebenen Fall einer ösophagealen Metastase eines follikulären Schilddrüsenkarzinoms, welche zwei Jahre nach operativer Schilddrüsenentfernung als mukosale, polypoide Metastase im Ösophagus auftrat. Die Metastase bei diesem Patienten wurde damals endoskopisch entfernt und der Patient laborchemisch nachkontrolliert.
Das follikuläre Schilddrüsenkarzinom metastasiert in der Regel eher in die Lungen, die Knochen und das Gehirn (1). Das Gesamtüberleben von kurativ behandelten, mittelalten Patienten mit Schilddrüsenkarzinomen ist ungefähr 80–95 % (2).
Einen ebenfalls ähnlichen Fall beschreiben die Kollegen Akyildiz et al. 2005 (3). Hier wird über ein Hürthle-Zell-Karzinom im Ösophagus berichtet, welches acht Jahre nach subtotaler Thyreoidektomie wegen multipler Knoten in der Schilddrüse auftrat. In diesem Fall wuchs der Tumor jedoch per continuitatem in den Ösophagus hinein. Auch in dieser Arbeit wurde der initiale Befund submukosal im Ösophagus als Hämangiom interpretiert, wie auch in unserem Fall, wo vor neun Jahren die Endosonografie den Verdacht auf ein Gefässkonvolut ergab.
In einem schönen Review aus dem Jahr 2001 berichten E. J. Simchuk, D. E. Low et al. (4) über sechs Fälle von Metastasen im Ösophagus, ausgehend von extraösophagealen Tumoren. Das mittlere Alter der Patienten in der Arbeit lag bei 72 Jahren (Range 68 ± 74). Zwei der primären Tumoren waren Lungentumore, vier waren Brustkrebs. Das durchschnittliche Intervall der Diagnose der Metastase zur Diagnose des Primarius lag bei sieben Jahren bei Patienten mit Brustkrebs, und bei fünf Monaten bei Patienten mit Lungenkrebs. Drei der Patienten wurden aufgrund des stenosierenden Verlaufs der Metastasierung mittels Stenting versorgt, und drei der Patienten starben aufgrund von gastrointestinalen Blutungen.
Autopsiestudien vermuten die Inzidenz von Metastasen im Ösophagus bei Patienten, die aufgrund irgendeiner Krebsart sterben um ca. 3–6 %, wobei Brust- und Lungenkarzinome die häufigsten Primärtumore zu sein scheinen (4). Der erste Fallbeschrieb einer ösophagealen Metastase wurde im Jahr 1942 veröffentlich, wobei hier als Primärtumor ein Prostatakarzinom vorlag (5).
Die Mitbeteiligung des Ösophagus im Rahmen eines anderen Primarius geschieht durch drei verschiedene Mechanismen, wobei der häufigste Mechanismus die direkte Infiltration von benachbarten Organen ist, namentlich Larynx, Hypopharynx, Trachea, Bronchus, Magen und mediastinale Lymphknotenmetastasen. Die anderen beiden Mechanismen sind die lymphogene Metastasierung und die hämatogene Metastasierung. Die Schwierigkeit der Detektion der Metastasen liegt darin, dass die Metastasierung häufig submukosal geschieht und der endoskopische Befund durch die normale darüberliegende Schleimhaut maskiert wird (4).
In dem von uns beschriebenen Fall lag die Zeit zwischen Erstdiagnose des Primarius und Diagnose der symptomatischen Metastase bei 27 Jahren.
Key Messages
• Zur Abklärung einer möglichen gastrointestinalen Blutung gehört die Endoskopie des oberen Gastrointestinaltraktes und bei fehlenden Erklärungen dann auch die des unteren respektive auch des mittleren Magendarmtraktes.
• Die Vorgeschichte des Patienten respektive die Kenntnis um die Vorerkrankungen ist bei unklaren Fällen essenziell.
• Ein patientenorientierter Therapieansatz sollte gerade bei dem immer älter werdenden Patientengut angestrebt werden.
• Metastasen von anderen Primärtumoren im Ösophagus sind selten und stammen hauptsächlich aus Lungen- und Brusttumoren.
Die Autorin und die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
• Zur Abklärung einer möglichen gastrointestinalen Blutung gehört die Endoskopie des oberen Gastrointestinaltraktes und bei fehlenden Erklärungen dann auch die des unteren respektive auch des mittleren Magendarmtraktes.
• Die Vorgeschichte des Patienten respektive die Kenntnis um die Vorerkrankungen ist bei unklaren Fällen essenziell.
• Ein patientenorientierter Therapieansatz sollte gerade bei dem immer älter werdenden Patientengut angestrebt werden.
• Metastasen von anderen Primärtumoren im Ösophagus sind selten und stammen hauptsächlich aus Lungen- und Brusttumoren.
1. Belinda Lee, Gary Cook, Lynn John, Kevin Harrington and Chris Nutting. Follicular Thyroid Carcinoma Metastasis to the Esophagus Detected by 18FDG PET/CT. Thyroid 2008. Volume 18; 267-271
2. M J Schlumberger. Papillary and Follicular Thyroid Carcinoma. N Engl J Med 1998; 338:297-306.
3. Murat Akyildiz, Omer Ozutemiz, Fulya Gunsar et al. Esophageal Metastasis Of Hurthle Cell Thyroid Carcinoma Eight Years After A Subtotal Thyroidectomy That Mimicked Esophageal Hemangioma. J Gastroenterol Hepatol . 2005 Oct;20(10):1628-9.
4. E J Simchuk, D E Low. Direct esophageal metastasis from a distant primary tumor is a submucosal process: a review of six cases. Dis Esophagus . 2001;14(3-4):247-50
5. Gross P, Freedman LJ. Obstructing secondary carcinoma of the esophagus. Arch Pathol Lab Med 33; 361-364: 1942
Nachdem eine junge Patientin eine intravenöse Eisensubstitution bei schwerer symptomatischer Eisenmangelanämie erhalten hatte, wurde eine schwere Hypophosphatämie mit begleitend leichtgradiger Muskelschwäche diagnostiziert. Bei renalem Phosphatverlust und erhöhtem Fibroblast Growth Factor 23 (FGF23) im Serum wurde die Diagnose einer Hypophosphatämie nach Verabreichung von Eisencarboxymaltose (Ferinject®) gestellt. Durch Hochregulation von FGF23 wird die renale Phosphatrückresorption gehemmt und die Aktivierung von Vitamin-D reduziert, wodurch die intestinale Phosphataufnahme weiter vermindert wird. Die Symptome einer Hypophosphatämie können nach erfolgter Eisensubstitution maskiert sein.
Eine 29-jährige, bislang gesunde Frau ohne vorbestehende Dauermedikation stellte sich mit ausgeprägter Schwäche, Müdigkeit und Belastungsdyspnoe seit einigen Wochen auf der Notfallstation vor. Die Diagnose einer schweren Eisenmangelanämie mit einem Hämoglobin (Hb) von 68 g/l (Normwert 120–160 g/L) wurde gestellt (Tab. 1), es erfolgte eine intravenöse (i. v.) Substitution mit 1000 mg Eisencarboxymaltose (Ferinject®).
In der ersten Verlaufskontrolle nach drei Wochen berichtete die Patientin über ein gesteigertes Wohlbefinden sowie verringerte Müdigkeit. Das Hb stieg auf 108 g/l. Auffallend war jedoch ein Serumphosphat unterhalb der Nachweisgrenze (< 0.32 mmol/l, Normwert 0.8–1.5 mmol/l). Eine leichtgradige beinbetonte Muskelschwäche wurde auf Nachfrage beklagt. Wir vermuteten eine Maskierung der Symptome einer schweren Hypophosphatämie bei kurzfristig gebesserten Anämiebeschwerden.
Die renale Phosphatexkretion war mit 44 % (Normwert 5–20 %, bei Hypophosphatämie < 5 %) deutlich erhöht. Zudem lag ein Vitamin-D-Mangel vor, und das Parathormon (PTH) und der Fibroblast Growth Factor 23 (FGF23) waren erhöht.
Differenzialdiagnostische Überlegungen
Eine Hypophosphatämie kann verschiedene Ursachen haben. Entstehen kann sie beispielsweise durch eine verminderte intestinale Aufnahme, eine erhöhte renale Ausscheidung oder durch einen Phosphat-Shift von extra- nach intrazellulär – letzteres bei stimulierter Glykolyse im Rahmen eines Refeeding-Syndroms oder einer respiratorischen Alkalose (1). Ebenso ist eine Hypophosphatämie bei Nierenersatzverfahren möglich (2).
Eine verminderte intestinale Phosphataufnahme war bei unserer Patientin denkbar, da sie phasenweise nur eine Mahlzeit pro Tag einnahm. Zudem kann ein Vitamin-D-Mangel, welcher auch bei unserer Patientin bestand, zusätzlich eine verminderte intestinale Phosphataufnahme bewirken (1).
Fastenperioden führen jedoch selten zu einer solch schweren isolierten Hypophosphatämie (1). Auch die übrigen unauffälligen Elektrolyte und die normwertigen Spiegel von Vitamin B12, Albumin und Folsäure bei unserer Patientin sprachen gegen das Vorliegen einer relevanten Mangelernährung.
Bei erhöhter Phosphatexkretion im Urin dachten wir differenzialdiagnostisch an einen renalen Phosphatverlust. Ein Hyperparathyreoidismus, welcher bei unserer Patientin vorlag, sorgt für eine verminderte renale Phosphatrückresorption und führt somit zu einer erhöhten renalen Phosphatausscheidung. Bei stets normwertigem Serumcalcium war ein primärer Hyperparathyreoidismus jedoch wenig wahrscheinlich. Wir werteten deshalb das erhöhte PTH am ehesten im Rahmen eines sekundären Hyperparathyreoidismus als Folge des Vitamin-D-Mangels. Eine weitere Differenzialdiagnose für einen erhöhten renalen Phosphatverlust bei erhöhtem FGF23 ist eine unerwünschte Arzneimittelwirkung nach I.-v.-Eisensubstitution (3–5).
Weitere Abklärungsschritte und Verlauf
Aufgrund der milden Symptomatik entschieden wir uns, trotz der schweren Hypophosphatämie, für eine orale Substitution mit Reducto®-spezial (Kaliumdihydrogenphosphat und Natriummonohydrogenphosphat-Dihydrat, insgesamt 3.6 g Phosphat täglich). Infolge einer allergischen Hautreaktion mussten wir auf Kaliumphosphat 1 molar B. Braun®-Ampullen zur oralen Einnahme wechseln. Darunter konnte über mehrere Wochen eine schrittweise Erhöhung des Serumphosphates erreicht und die Phosphatsubstitution stufenweise reduziert werden. Begleitend erfolgte eine orale Vitamin-D-Substitution.
Nach insgesamt sechs Wochen zeigte sich das Serumphosphat unter gestoppter oraler Phosphateinnahme anhaltend im Normbereich. Anamnestisch war bereits wenige Tage nach Therapiebeginn keine Muskelschwäche mehr auszumachen.
Sechs Monate später präsentierte sich die Patientin erneut mit einer Eisenmangelanämie bei einem Hb von 79 g/l trotz hochdosierter oraler Eisensubstitution. Eine gynäkologische Blutungsquelle bei Uterus myomatosus wurde vermutet. Wir entschieden uns für eine erneute Eiseninfusion. Bei Status nach Hypophosphatämie nach Eisencarboxymaltose verabreichten wir nun 1000 mg Eisenisomaltose (Monofer®).
Der Hb-Wert stieg im Verlauf auf 100 g/l an. Laborchemisch war in den Tagen danach erneut eine moderate Hypophosphatämie (Phosphat minimal 0.43 mmol/l) zu sehen, klinisch mit leichter Muskelschwäche einhergehend. Es erfolgte eine passagere orale Phosphatsubstitution (Tab. 2).
Diagnose
Wir stellten die Diagnose einer schweren Hypophosphatämie als unerwünschte Arzneimittelwirkung nach intravenöser Eisensubstitution. Passend dazu liess sich nach der Verabreichung von Eisencarboxymaltose (Ferinject®) eine neue Hypophosphatämie detektieren. Zudem fand sich eine deutliche FGF23-Erhöhung im Serum sowie eine erhöhte renale Phosphatexkretion. Die erhöhte FGF23-Konzentration im Blut hemmte die tubuläre Phosphatrückresorption und führte somit zu einer erhöhten renalen Phosphatausscheidung (3, 6). Der zugrunde liegende Mechanismus für den Anstieg von FGF23 konnte bislang nicht vollständig geklärt werden. Wolf et al. vermuten, dass Eisenpräparate den FGF23-Abbau durch Osteozyten hemmen und somit mehr aktives FGF23 anfällt (6).
Eine erhöhte FGF23-Konzentration führt auch zu einer verminderten Umwandlung von 25-OH-Vitamin-D in 1.25-OH-Vitamin-D (Calcitriol). Ein Vitamin-D-Mangel seinerseits vermindert die intestinale Calciumresorption, wodurch es zu einer erhöhten Freisetzung von PTH kommt (3). PTH reduziert die renale und ein Calcitriol-Mangel die intestinale Phosphatresorption. So wird die Hypophosphatämie weiter verstärkt (1).
Kommentar
Die Häufigkeit einer Hypophosphatämie nach Eisensubstitution variiert je nach Präparat.
Wolf et al. beobachteten, dass es bei Eisencarboxymaltose etwa 9-mal häufiger zu einer schweren Hypophosphatämie (Serum-Phosphat ≤ 1 mg/dl [entspricht 0.32 mmol/l, Anmerkung Autorin]) kommt verglichen zu Eisenisomaltose (7).
Schaefer et al. beschrieben ein bis zu 20-fach erhöhtes Risiko für eine Hypophosphatämie durch Eisencarboxymaltose verglichen zu Eisenisomaltose. In derselben Studie wurden schwere Hypophosphatämien nur in der Gruppe mit Eisencarboxymaltose verzeichnet (8).
Unsere Patientin erhielt initial 1000 mg Eisencarboxymaltose (Ferinject®). Das Serumphosphat lag am Tag der Verabreichung im Normbereich. Drei Wochen später zeigte sich ein schwerer Phosphatmangel.
Auch nach Verabreichung von 1000 mg Eisenisomaltose (Monofer®) trat eine substitutionsbedürftige Hypophosphatämie auf. Dies verdeutlicht, dass es auch beim Einsatz von Eisenisomaltose zu einer Hypophosphatämie kommen kann, trotz geringeren Risikos.
Eine Hypophosphatämie kann bereits nach einmaliger Eiseninfusion auftreten (8). Die Patientinnen und Patienten sollten vorgängig über die Symptome eines Phosphatmangels aufgeklärt werden. Diese umfassen Muskelschwäche, Schluckstörungen, gastrointestinale Beschwerden (Ileus) sowie Atemnot als Ausdruck einer Herz- oder Ateminsuffizienz. Neurologisch sind Parästhesien, eine verstärkte Reizbarkeit und Verwirrtheit beschrieben. Bei länger andauernden Hypophosphatämien kann auch der Mineral- und Knochenhaushalt gestört sein (1, 8).
Dieses Fallbeispiel zeigt, dass auch bei leichtgradiger Symptomatik eine schwere Hypophosphatämie vorliegen kann. Hierbei ist eine zusätzliche Beschwerdemaskierung durch die Korrektur eines Eisenmangels möglich. Therapeutisch sollte bei leichten Symptomen primär eine orale Phosphatsubstitution erfolgen, ein begleitender Vitamin-D-Mangel muss berücksichtigt und ebenfalls ausgeglichen werden.
Abkürzungen FGF23 Fibroblast Growth Factor 23 Hb Hämoglobin i. v. Intravenös PTH Parathormon
Dr. med. Laura Giezendanner
Klinik für Allgemeine Innere Medizin/Hausarztmedizin und Notfallmedizin
Kantonsspital St. Gallen
Laura.Giezendanner@kssg.ch
Dr. med. univ. Franziska Vogler
Klinik für Allgemeine Innere Medizin/Hausarztmedizin
und Notfallmedizin, Kantonsspital St. Gallen
Rorschacherstrasse 95,
9007 St. Gallen
Dr. med. Andrea Hausammann
Oberärztin mbF
Klinik für Allgemeine Innere Medizin/Hausarztmedizin und Notfallmedizin
Kantonsspital St. Gallen
Die Autorinnen haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Gaasbeek A, Meinders AE. Hypophosphatemia: an update on its etiology and treatment. Am J Med 2005;118:1094–101. https://doi.org/10.1016/j.amjmed.2005.02.014.
2. Broman M, Carlsson O, Friberg H, Wieslander A, Godaly G. Phosphate-containing dialysis solution prevents hypophosphatemia during continuous renal replacement therapy. Acta Anaesthesiol Scand 2011;55:39–45. https://doi.org/10.1111/j.1399-6576.2010.02338.x.
3. Schaefer B, Tobiasch M, Wagner S, Glodny B, Tilg H, Wolf M, et al. Hypophosphatemia after intravenous iron therapy: Comprehensive review of clinical findings and recommendations for management. Bone 2022;154:116202. https://doi.org/10.1016/j.bone.2021.116202.
4. Wolf M, Chertow GM, Macdougall IC, Kaper R, Krop J, Strauss W. Randomized trial of intravenous iron-induced hypophosphatemia. JCI Insight 2018;3:e124486. https://doi.org/10.1172/jci.insight.124486.
5. Schaefer B, Meindl E, Wagner S, Tilg H, Zoller H. Intravenous iron supplementation therapy. Mol Aspects Med 2020;75:100862. https://doi.org/10.1016/j.mam.2020.100862.
6. Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women: FGF23 IN IRON DEFICIENCY. J Bone Miner Res 2013;28:1793–803. https://doi.org/10.1002/jbmr.1923.
7. Wolf M, Rubin J, Achebe M, Econs MJ, Peacock M, Imel EA, et al. Effects of Iron Isomaltoside vs Ferric Carboxymaltose on Hypophosphatemia in Iron-Deficiency Anemia: Two Randomized Clinical Trials. JAMA 2020;323:432. https://doi.org/10.1001/jama.2019.22450.
8. Schaefer B, Würtinger P, Finkenstedt A, Braithwaite V, Viveiros A, Effenberger M, et al. Choice of High-Dose Intravenous Iron Preparation Determines Hypophosphatemia Risk. PLOS ONE 2016;11:e0167146. https://doi.org/10.1371/journal.pone.0167146.
9. Hardy S, Vandemergel X. Intravenous Iron Administration and Hypophosphatemia in Clinical Practice. Int J Rheumatol 2015;2015:1–6. https://doi.org/10.1155/2015/468675.
Die thrombotische Mikroangiopathie (TMA) wird durch die typische Trias einer schweren Thrombozytopenie, Coombs-negativen hämolytischen Anämie sowie Endorgandysfunktion definiert. Pathophysiologisch handelt es sich um Ischämie-bedingende Mikrothromben in Arteriolen und Kapillaren, welche zu schwerwiegender Organdysfunktion sowie akut lebensbedrohlichen Endorganschädigungen führen können. Hinsichtlich Ätiologie, Verlauf, Therapie und Prognose werden die folgenden Manifestationsformen unterschieden: die thrombotisch-thrombozytopenische Purpura (TTP), das Shigatoxin-induzierte hämolytisch-urämische Syndrom (STEC-HUS), die sekundäre TMA und das atypische hämolytisch-urämische Syndrom (aHUS).
Wir präsentieren den Fall eines 49-jährigen lungentransplantierten Patienten mit aHUS. Die Komplexität der zugrunde liegenden Pathomechanismen der TMA, die schwierige Differenzierung der TMA-Manifestationen und das anspruchsvolle Management eines aHUS nach Lungentransplantation verdeutlichen die Einzigartigkeit dieses Patientenfalles.
Die TTP resultiert aus einer genetisch bedingten oder erworbenen Reduktion der Aktivität der von-Willebrand-Faktor (vWF) spaltenden Metalloproteinase ADAMTS13. Bei der selteneren kongenitalen TTP kommt es mutationsbedingt zu einer verringerten hepatischen Produktion von ADAMTS13. Die deutlich häufigere erworbene Form, auch «acquired TTP» (aTTP) genannt, betrifft vor allem junge Erwachsene ohne Vorerkrankungen und ist in einer IgG-Autoantikörper vermittelten Funktionseinschränkung des Enzyms begründet (1). Die aTTP, welche ätiologisch die Mehrheit aller TTP-Fälle ausmacht, gehört zur Gruppe seltener Erkrankungen mit einer jährlichen Inzidenz von 1.5 bis 6 Fällen pro 1 Million Einwohner in Europa (2, 3). Der Defekt der Protease resultiert in einer Multimerbildung des vWF-Glykoproteins.
Diese bedingen eine aggravierte Thrombozytenaggregation mit folgender mikrovaskulärer Thrombosierung und Hämolyse (4). Diagnostisch wegweisend ist die klinische Präsentation einer renalen Dysfunktion mit begleitenden neurologischen Symptomen in Kombination mit Thrombozytopenie und hämolytischer Anämie (4, 5). Im Falle der sekundären TMA ist die Ätiologie vielseitig; beschriebene Auslöser reichen von bakteriellen, viralen und fungale Infektionen, Krebserkrankungen, Autoimmunerkrankungen (z. B. systemischer Lupus Erythematodes, systemische Sklerose, Antiphospholipid- Antikörper-Syndrom), Organ- oder Knochenmarktransplantationen und Schwangerschaft bis zu Drogenkonsum sowie medikamentösen Triggern (4). Fall-Kontroll-Studien aus dem angloamerikanischem Raum beschreiben maligne Erkrankungen als die häufigsten Risikofaktoren einer sekundären TMA (2). Im Falle einer nicht ADAMTS13-Aktivität bedingten TMA führt eine systemische Endothelschädigung durch immunzelluläre und Komplement-abhängige Pathomechanismen zur mikrovaskulären Thrombosierung.
Das STEC-HUS, auch typisches HUS genannt, ist eine Komplikation im Rahmen einer gastrointestinalen Infektion mit Shigatoxin-bildenden Erregern – meist enterohämorrhagischen Escherichia coli. Das STEC-HUS tritt typischerweise bei Kindern im Alter zwischen zwei und fünf Jahren auf, kann allerdings im Rahmen von epidemischen Ausbrüchen auch andere Altersgruppen betreffen (6). Die Zerstörung renaler und intestinaler Endothelzellen ergibt das klinische Bild von meist blutigen Durchfällen und akuter Nierenfunktionseinschränkung (AKI).
Im Falle des atypischen HUS (aHUS) besteht eine endogene Prädisposition für ein hyperreagibles Komplementsystem. Zeitpunkt und Schwere der klinischen Manifestation sind höchst individuell und meist mit dem Auftreten endo- beziehungsweise exogener Stressoren assoziiert, sogenannter Komplement-aggravierender Faktoren. Ursächlich ist ein genetisch bedingter Funktionsverlust von Komplementregulatoren, das Vorliegen von Gain-of-function-Mutationen in Komplement-codierenden Genabschnitten oder eine Autoantikörper bedingte Hemmung von Komplementinhibitoren (4). Obwohl das aHUS als primäre Erkrankung des Komplementsystems definiert ist, handelt es sich im klinischen Alltag oft um eine differenzialdiagnostische Ausschlussdiagnose. Das heisst, es gibt keinen laborchemischen Test, welcher die Erkrankung im akuten Setting eindeutig diagnostizieren kann. Sie wird als «ultra-rare-disease» eingestuft, mit einer geschätzten jährlichen Inzidenz weltweit von 0.23–1.9/Million (7, 8, 9). Die Prognose ist mit einer Mortalität von 25 % und einem Patientenanteil von bis zu 50 %, der in der Akutphase ein Nierenversagen entwickelt, deutlich schlechter als beim typischen HUS, welches mit einer Letalität von 1–5 % die beste Prognose aller TMA-Manifestationen besitzt (10–12).
Fallbericht
Zum ersten Mal vorstellig wurde unser Patient im Oktober 2021 mit pulmonal rasch progredienter Symptomatik im Sinne einer connective tissue disease-associated interstitial lung disease (CTD-ILD) bei neu diagnostiziertem Sjögren- Syndrom. Der Patient präsentierte eine rasche lungenfunktionelle Verschlechterung im Sinne einer progredienten restriktiven Ventilations- und Diffusionsstörung, die innerhalb weniger Monate zu einer respiratorischen Insuffizienz führten. Dank frühzeitiger Zuweisung konnte der Patient nach einer zweiwöchigen Lungentransplantationsabklärung im Universitätsspital Zürich im Oktober 2022 bilateral lungentransplantiert werden.
Ende April 2023 stellte sich der Patient mit Thoraxschmerzen, Kopfschmerzen und Verschlechterung des Allgemeinzustands vor. Es präsentierte sich eine Panzytopenie mit aggravierter Thrombozytopenie (Nadir 12 g/l), deutlich erhöhter LDH, erhöhtem Bilirubin, einer transfusionspflichtigen hämolytischen Anämie (Hb 51 g/l) und rapider Verschlechterung der Nierenfunktion im Rahmen einer akuten Nierenfunktionseinschränkung Stufe 3 nach KDIGO. Die mikroskopische Zelldifferenzierung zeigt 20–25 Fragmentozyten pro Gesichtsfeld im mikroskopisch hypochrom-anisozytären Blutbild. Der direkte Antiglobulintest (Coombs-Test) zeigte sich negativ (DAT-). Die Parameter Folsäure, Vitamin B12 und Ferritin befanden sich im Normbereich. Eine Neutropenie war bekannt und wurde im Rahmen der knochenmarksuppressiven Medikation des lungentransplantierten Patienten (Ganciclovir, Itraconazol, Mycophenolat mofetil [MMF]) interpretiert. Zum Zeitpunkt der Vorstellung war der Patient aufgrund erhöhter Donor-spezifischen Antikörpern im Serum mit Tacrolimus, MMF, dem mTOR-Inhibitor Everolimus und Prednison vierfach immunsupprimiert.
In der Zusammenschau konnte bei obig bereits beschriebener Thrombozytopenie, transfusionspflichtiger hämolytischer Anämie und rapider Verschlechterung der Nierenfunktion noch am Vorstellungstag die klinische Verdachtsdiagnose einer TMA gestellt werden. Bei annähernd normwertiger ADAMTS13-Aktivität (45 %) sowie negativer EHEC-/Shigatoxin-Testung im Stuhl konnten eine TTP sowie ein STEC-HUS schnell ausgeschlossen werden (Abb. 1).
Es ergaben sich die Differenzialdiagnosen einer sekundären medikamentösen TMA, am ehesten unter kombinierter Calcineurin- und mTOR-Inhibitor-Therapie oder eines atypischen HUS. Um eine mögliche medikamentöse Ursache zu adressieren, wurde ein Calcineurin-Inhibitor-Wechsel innerhalb der Substanzklasse (Tacrolimus zu Ciclosporin A) vorgenommen sowie Everolimus pausiert. Zur weiteren Evaluation einer Komplement-assoziierten TMA im Rahmen eines aHUS wurde die Konzentrationsbestimmung der Komplementfaktoren vorgenommen. Eine Erhöhung von Komplement CD5b-9 MAC konnte bestätigt werden. C3, C4, Faktor H und I waren hingegen unauffällig.
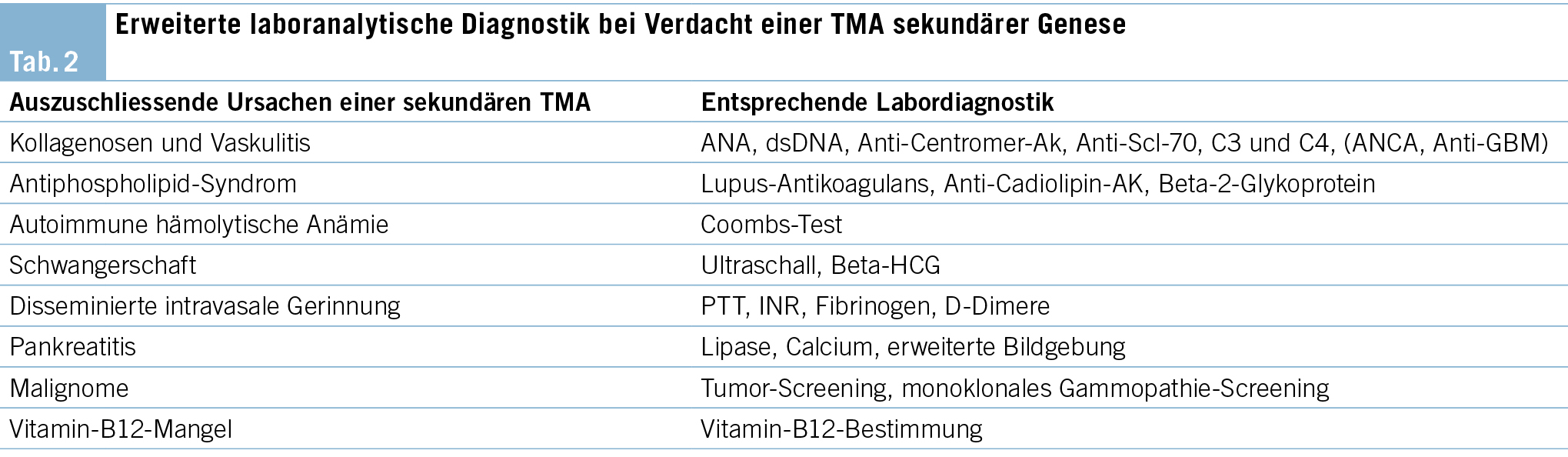
Die vaskulitische Assoziation der hier als Grundmorbidität vorliegende Kollagenose eines Sjögren-Syndroms spielt als autoimmunologischer, prädisponierender Faktor ebenfalls eine Rolle im diagnostischen Abklärungsprozess bei Verdacht einer sekundären TMA (Tab. 1), insbesondere im Falle des hier am ehesten vorliegenden aHUS.
Es erfolgte eine molekulargenetische Diagnostik zur Identifizierung pathogener Mutationen in Komplement-assoziierten Genen (Next Generation Sequencing panel aHUS). Es lag eine Duplikation der im Tandem auf Chromosom 1q lokalisierten mit aHUS assoziierten Gene CD46, CFH und CFHR1–5 vor. Eine Knochenmarkpunktion zeigte lediglich hyporegeneratorische Zellreihen ohne Malignitätshinweise und verblieb somit ohne Hinweis auf ein myeloproliferatives Syndrom.
Ergänzend erfolgte eine Nierenbiopsie, deren Befund mit einem aHUS vereinbar war. Nach interdisziplinärer Rücksprache wurde umgehend mit einer empirischen anti-komplementären Therapie mit Eculizumab begonnen. Unter bestehender Immunsuppression mittels Ciclosporin, MMF und Prednison nach Transplantation wurde unser Patient vor Beginn der Eculizumab-Therapie gegen Meningokokken und Pneumokokken geimpft. Da bei Triple-Immunsuppression nur eine geringe Impfantwort zu erwarten ist, wurde eine antiinfektive Meningokokkenprophylaxe mit Ciprofloxacin 500mg/d begonnen, die bis mindestens sechs Monate nach letzter Eculizumab-Gabe fortgeführt wurde.
Trotz initial raschen Ansprechens – demonstriert durch steigende Thrombozytenwerte, sich stabilisierendem Hämoglobin sowie einer verbesserten Nierenfunktion – musste das Arzneimittel aufgrund ungeklärter finanzieller Kostenübernahme nach lediglich vier Wochen erfolgter Therapie pausiert werden. Nach zwei Wochen Therapiepause kam es zum TMA-Rezidiv mit erneut fallenden Thrombozyten und Erythrozytenzahlen sowie Verschlechterung der Nierenfunktion.
Das klinische Ansprechen auf Eculizumab konnte nach Wiederbeginn der Therapie gänzlich objektiviert werden (Diagnosis ex juvantibus). Es kam erneut zu einer Stabilisierung der Nierenwerte und der Hämatologie. Die rezidivierende Messung der Komplementaktivität und von C5b-9 zeigte eine gute Suppression des Komplementverbrauchs auf dem alternativen Komplementweg unter der Antikörpertherapie.
Diagnostik
Die Verdachtsdiagnose einer TMA wird bei einer absoluten oder relativen Thrombozytopenie (Thrombozyten < 150 G/l, > 25 % Reduktion von der Baseline) in Kombination mit einer Coombs-negativen hämolytischen Anämie mit Fragmentozyten im Blutausstrich, erhöhter LDH, erhöhtem Bilirubin, reduziertem Haptoglobin und Retikulozytose (kann bei Nierenversagen fehlen) sowie Endorgandysfunktion gestellt (Tab. 1). Entscheidend ist nach Diagnose einer TMA, im Anschluss die Ätiologie korrekt zuordnen zu können, um eine zielgerichtete Therapie zu ermöglichen, da sich die Therapie je nach Ätiologie erheblich unterscheidet. Für das optimale Management der mitunter lebensbedrohlichen Organmanifestationen der TMA ist eine kontinuierliche, interdisziplinäre Zusammenarbeit zwischen Nephrologen, Hämatologen, Neurologen und Genetikern essenziell.
Eine frühzeitige Bestimmung der ADAMTS13-Aktivität hat das Ziel, eine TTP als Ursache rasch auszuschliessen. Es folgt die Beurteilung etwaiger infektiöser Ätiologien mit der Durchführung von Real-Time PCR/ELISA zur Testung auf Shigatoxin im Stuhl oder einem Analabstrich sowie einer Röntgen-Thorax-Aufnahme, der Abnahme von Blut-, Sputum, Stuhl- und Urinkulturen, einem Rachenabstrich auf Influenza, Testung auf Pneumokokken-Ag im Urin sowie dem Ausschluss von HIV, Hepatitis B und C. Weiterhin sollten relevante Komorbiditäten und Medikamente als mögliche Auslöser in einer detaillierten Anamnese erfasst werden. Die in Tab. 2 aufgeführten Analysen gelten lediglich als Startpunkt der erweiterten Abklärung und müssen je nach klinischem Verdacht gezielt erweitert werden.
Eine Nierenbiopsie wird nicht nur zur Bestätigung einer TMA als Ursache der Nierenschädigung herangezogen, sondern erlaubt auch die Einschätzung, inwiefern bereits ein chronischer Schaden besteht, und ermöglicht somit eine Aussage über die Prognose. Manchmal kann sie auch Hinweise auf die Ursache der TMA liefern. In der klinischen Praxis wird eine Nierenbiopsie aufgrund der Thrombozytopenie und dem damit einhergehenden Blutungsrisiko initial oft verzögert durchgeführt.
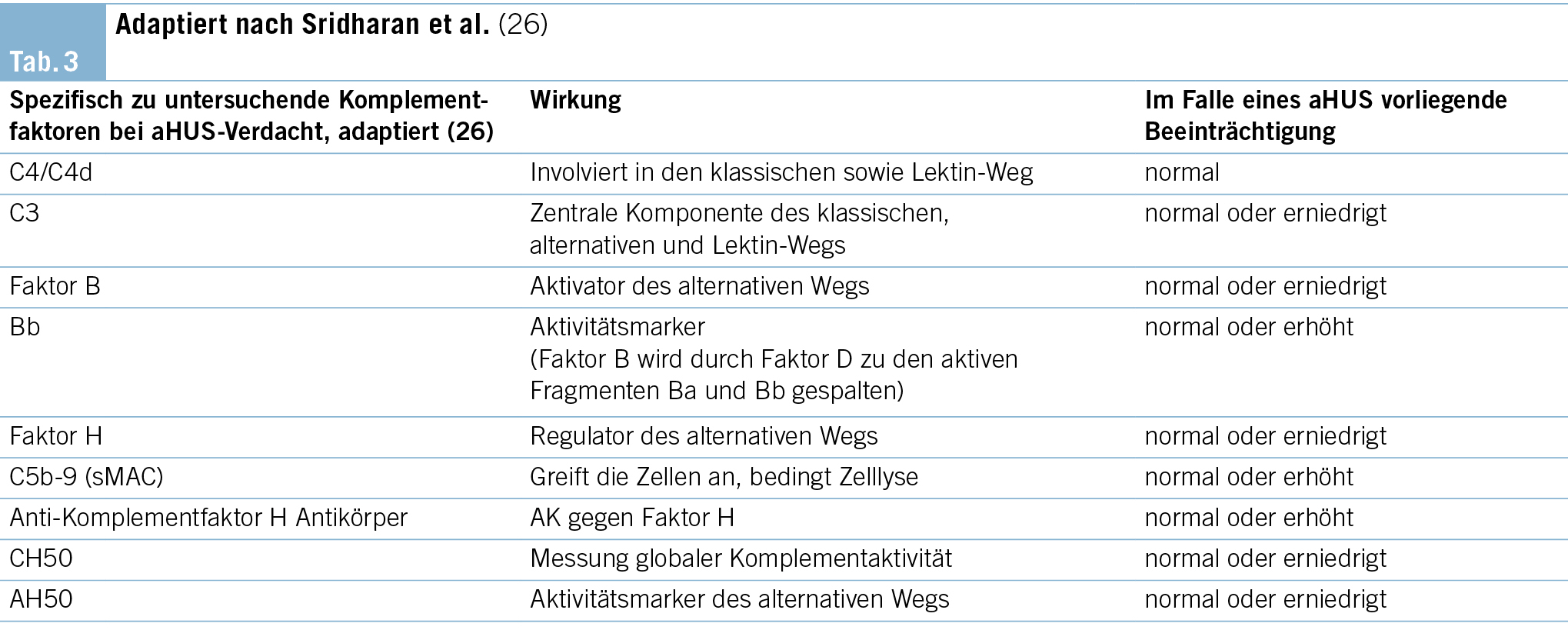
Zeigen die genannten Untersuchungen unauffällige Befunde an, sollte eine Komplementfaktoraktivitätsprüfung kombiniert mit einer genetischen Analyse erfolgen. Bei aHUS-Patienten kann bei ungefähr 40–60 % aller Patienten eine genetische Veränderung im Bereich der Komplementkaskade nachgewiesen werden (13). Tab. 3 führt die wichtigsten Komplementfaktoren, Komplementantikörper und Komplement-codierenden Gene zur Testung auf. Die Bestimmung von Komplementfaktoren und die Sequenzierung von Risikogenen hilft jedoch nicht bei der initialen Diagnosestellung eines aHUS, respektive bei der Therapieentscheidung, ob eine Komplementblockade mit Eculizumab begonnen werden soll, da die Analysen längere Zeit in Anspruch nehmen.
Zum jetzigen Zeitpunkt sind Mutationen in mehr als zehn unterschiedlichen Komplement-codierenden Genen bekannt. Die häufigste und somit klinisch relevanteste Mutation betrifft den hepatisch synthetisierten Komplementfaktor H auf dem CFH-Gen. Weiterhin wichtig und als Hochrisikomutationen bekannt sind Gain-of-function-Mutationen in codierenden Abschnitten von den Komplement-Faktoren B (CFB) und C3 (13).
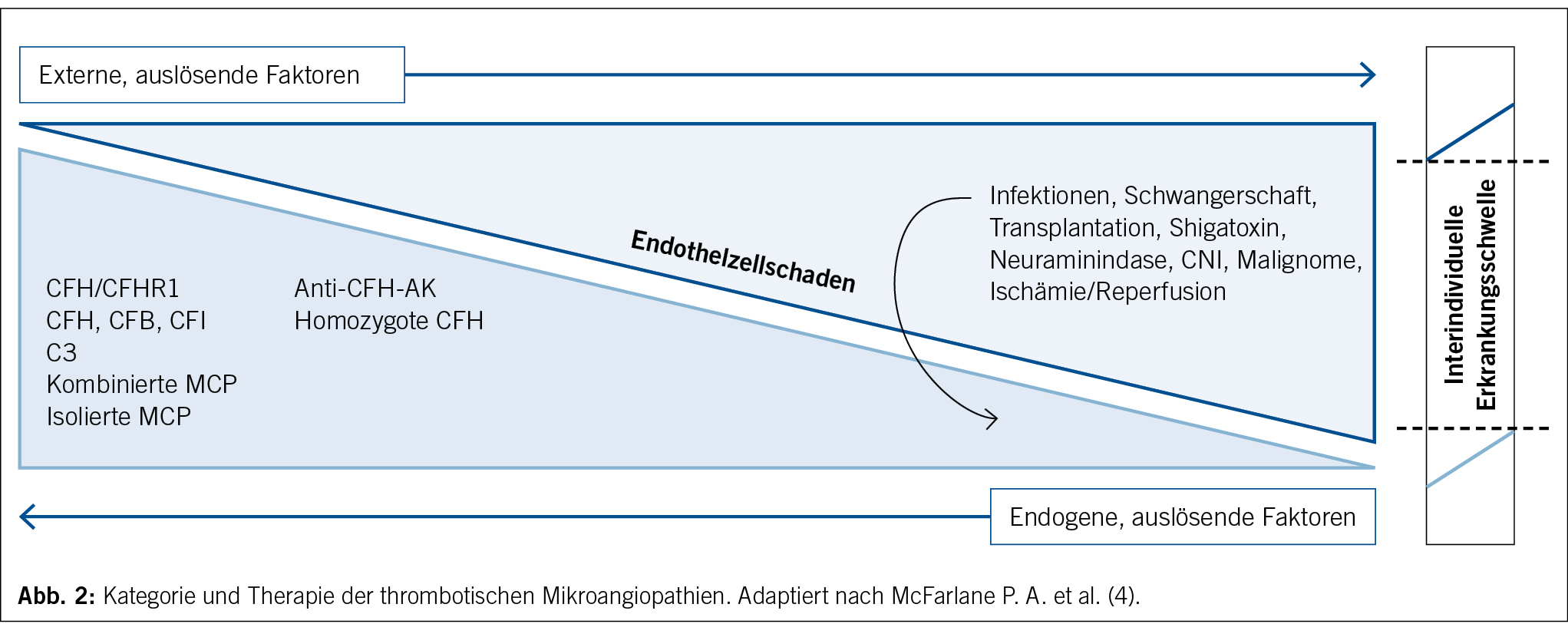
Wie bei vielen anderen hereditären Erkrankungen geht man auch bei der Pathogenese des aHUS von einer Two-Hit-Hypothese aus. Dies bedeutet, dass eine Genmutation allein nicht zwingend die Krankheit zur klinisch-apparenten Manifestation bringt. Studien haben gezeigt, dass selbst bei einer schweren CFH-Mutation die Penetranz bei unter 50 % liegt (13). Komplement-aggravierende Faktoren führen zur Überschreitung der interindividuellen Belastungsschwelle und folglich der Aktivierung des Komplementsystems bei bereits minimalen ersten Endothelschäden (Abb. 2) (13). Bei zugrunde liegender genetischer Prädisposition entwickelt sich daraus eine unkontrollierte, überschiessende systemische Komplementreaktion, welche die fulminante Manifestation der Erkrankung mit folgenden Endorganschäden bedingt (13) (14). Die daraus resultierende Hämolyse aggraviert das überaktive Komplementsystem weiter, sodass ein sich selbst unterhaltender Kreislauf entsteht, welcher den für die Erkrankung so typischen schwerwiegenden klinischen Verlauf bedingt. Weitere Charakteristika eines aHUS, die eine gewisse Abgrenzung zu anderen TMA-Manifestationen erlauben, sind ein familiär gehäuftes Auftreten und ein beobachtetes Rezidiv der Erkrankung nach Transplantation (4).
Therapieansätze
Ursprünglich bestand für Patienten mit TMA als einzige Therapieoption, nebst supportiven Massnahmen, der wiederholte Austausch des Blutplasmas (Plasmapherese). Für Patienten mit aHUS konnte dieser Ansatz selten einen ausreichenden therapeutischen Erfolg erzielen, da die zugrunde liegende Pathogenese nicht behoben wurde. Vor Einführung von komplementinhibierenden Substanzen wurden bis zu 40 % der Patienten trotz Plasmaaustausch oder sonstiger supportiver Therapie bereits mit der ersten klinischen Manifestation dialysebedürftig oder verstarben (10) (15). Im Fünf-Jahres-Verlauf betraf dies bis zu 64 % der adulten Patienten (1) (16).
Während der fulminanten Erstmanifestationsphase bis zum Erhalt der ADAMTS13-Bestimmung ist die Plasmapherese, heutzutage meist unterstützend mit Gabe von Fresh Frozen Plasma (FFP) (17) (Tab. 4), bei adulten Patienten mit TMA unklarer Ätiologie die Therapie der Wahl, da die Verzögerung der Therapie der TTP das klinische Outcome deutlich verschlechtert (4).
Im Falle einer bestätigten TTP besteht eine neue Therapieoption mittels Anti-vWF-Nanobody Caplacizumab. Es reduziert die Adhäsion zwischen grossen vWF-Multimeren und Blutplättchen und führt zu einer rascheren Normalisierung der Thrombozytenzahl, Reduktion der Therapiedauer sowie Wahrscheinlichkeit eines TTP-Rückfalls (17).
Sobald eine TTP ausgeschlossen werden kann und sekundäre Ursachen als unwahrscheinlich erachtet werden, ist mittlerweile die intravenöse Komplementinhibition mittels monoklonalen IgG-Antikörpern, Eculizumab oder Ravulizumab die kausale Therapie der Wahl einer Komplement-vermittelten TMA, da sie zur signifikanten Reduktion der renalen Endorganschädigung führt (18) (19) (20). Eculizumab und Ravulizumab binden mit hoher Affinität an C5 und verhindern so die Entstehung des terminalen Komplementkomplexes (Abb. 3).
Multiple Fallberichte sowie Phase-II-Studien haben bereits über die Wirksamkeit der terminalen Komplementblockade mittels Eculizumab berichtet, welches neben Ravulizumab der einzige aktuell in der Schweiz zugelassene Komplementinhibitor zur Therapie des aHUS ist (8). Ravulizumab besitzt eine längere Wirkdauer und wird bei Erwachsenen nach zwei Gaben im Abstand von zwei Wochen alle acht Wochen verabreicht, was für die Patienten häufig eine deutliche Vereinfachung der Therapie im Vergleich zu den zweiwöchentlichen Gaben von Eculizumab ist. Es ermöglicht somit eine Reduktion der durchschnittlichen jährlichen Infusionszeit und Behandlungsdauer auf höchstens sechs Stunden und somit deutlich mehr Flexibilität und ein Gewinn an Lebensqualität für die betroffenen Patienten (21). Therapieansprechen und -erfolg mit Senkung der Akutkomplikationen sowie das Verhindern einer chronischen Niereninsuffizienz sind jedoch massgeblich vom Zeitpunkt der korrekten Einordnung des Krankheitsbildes abhängig. Es besteht ein begrenztes therapeutisches Fenster, bevor irreversible renale Schäden eintreten (18).
Prognose
Eine Metaanalyse aus klinischen Fallberichten der letzten zehn Jahre zu Therapieansprechen bei aHUS konnte eine statistisch signifikante Reduktion der Mortalität durch die Gabe von Eculizumab zeigen (2.3 vs. 8.8 %, p = 0.045) (22). Nachweislich kann eine vollständige Hemmung der terminalen Komplementaktivität erzielt werden sowie der Erhalt beziehungsweise die Verbesserung der betroffenen Organfunktionen. Aktuell existiert keine klinische Leitlinie über die empfohlene Dauer der medikamentösen Komplementhemmung. Rezidive der Erkrankung sind insbesondere in den ersten Monaten nach Pausierung der Therapie häufig, können aber lebenslang auftreten. Beobachtungsstudien sowie das französische Nationalregister für aHUS-Erkrankungsfälle zeigten bei durchschnittlicher Therapiesistierung nach 18 Monaten Rückfälle bei bis zu 30 % der beobachteten Patienten (19) (23). Bei näherer Differenzierung der Patientengruppe konnte jedoch gezeigt werden, dass Eculizumab bei über 50 % der untersuchten Patienten erfolgreich sistiert wurde. Dies betraf insbesondere Patienten ohne identifizierbare Mutationen (13). Ein erhöhter CD5b-9-Plasmawert wiederum galt als Hinweis für ein erhöhtes Rezidivrisiko nach Absetzen der Medikation (24).
Diskussion
Der beschriebene Patientenfall präsentiert die Fallstricke und Herausforderungen im Rahmen von Diagnostik und Management einer thrombotischem Mikroangiopathie nach Organtransplantation. Das aHUS gilt als Ausschlussdiagnose, welche das interdisziplinäre Zusammenspiel verschiedener internistischer Disziplinen erforderlich macht. Vor allem die differenzialdiagnostische Unterscheidung einer sekundären TMA und eines aHUS stellt eine besondere Herausforderung dar. Der Kostenträger besteht auf einer klaren Unterscheidung der Entitäten, um die Kosten für die teure Komplement-inhibitorische Therapie im Falle eines aHUS zu übernehmen. Dies stellt sich als besonders schwierig heraus, da Auslöser einer sekundären TMA zugleich als aggravierende Faktoren der Komplementdysregulation im Rahmen eines aHUS fungieren können. So stellt eine Organtransplantation ein hohes Risiko für die Entstehung einer sekundären TMA als auch für die überschiessende Aktivierung eines dysregulierten Komplementsystems dar. In unserem Fall sind vor allem die immunsuppressiven Medikamente wie der mTOR-Inhibitor Everolimus und der Calcineurin-Inhibitor Tacrolimus zu nennen. Insbesondere die Kombination beider immunsuppressiven Arzneimittel nach Lungentransplantation bergen erwiesenermassen ein erhöhtes Risiko für die Entstehung einer TMA (25). Als ursächlich wird hier die Induktion der Thrombozytenaggregation durch die einzelnen Wirkstoffe sowie eine generelle endotheliale Zytotoxizität diskutiert, die in der Folge eine mikrovaskuläre Schädigung bedingen (25). Diese Medikamente sind nach Lungentransplantation für den Erhalt der Allograftfunktion lebenswichtig. Umso dramatischer stellt sich die Entscheidung dar, diese Medikamente zu reduzieren, zu pausieren oder zu wechseln. Bei dieser Unterscheidung helfen kann die genetische Testung auf eine zugrunde liegende Komplementmutation, welche der Auslöser einer aHUS-Manifestation sein kann.
Es ist an dieser Stelle wichtig zu bemerken, dass ein fehlender Mutationsnachweis der oben genannten Gene ein aHUS nicht ausschliesst, da bei einem grossen Teil der aHUS-Patienten keine spezifische Mutation nachgewiesen werden kann. Auch die Messung der Komplementaktivitätsmarker im Serum können ein aHUS nicht sicher von anderen TMA-Manifestationen unterscheiden. Zudem helfen die Resultate nicht bei der Entscheidung zur Therapieinitiierung, da hier schnell gehandelt werden muss, um eine irreversible Schädigung zu verhindern, und die Tests oft Tage bis Wochen (Komplementfaktoren) oder gar Monate benötigen (Komplementgenetik).
Sridharan et al. testeten zur Diagnose des aHUS neun verschiedene Komplementmarker (CH50, AH50, C3, C4, Faktor B, Faktor H, C4d, Bb sowie sC5b-9) (26). Obwohl die Sensitivität der Kombination dieser Marker annähernd 100 % darstellt, sollten dieselben aufgrund niedriger Spezifität nur in enger Zusammenschau der weiteren klinischen Befunde interpretiert werden. Die Kenntnis einer Komplement-aktivierenden Mutation ist essenziell hinsichtlich der Entscheidungen zur Therapiedauer (13). Aktuell besteht kein gemeinsamer Konsens über die zu empfehlende Dauer der Komplementinhibition bei aHUS. Während sich die Therapie als äusserst effektiv hinsichtlich Milderung der Krankheitsaktivität erwiesen hat, sind gleichzeitig, mitunter schwerwiegende Nebenwirkungen wie das Risiko einer Meningokokkenmeningitis und die hohen Kosten von bis zu 600 000 Euro pro Jahr und Patient zu bedenken (27) (28).
Eculizumab gilt als eines der teuersten Medikamente überhaupt auf dem pharmazeutischen Markt. Die Beobachtung, dass Rezidive nach Therapiestopp häufiger bei Patienten mit besonders hohen Spiegeln des terminalen Komplementkomplexes auftreten sowie dass ein positives Ergebnis der untersuchten prädisponierenden Gene im Sinne eines Mutationsnachweises ebenfalls ein erhöhtes Risiko für ein Rezidiv darstellt, verdeutlicht die Wichtigkeit der Genanalyse und zeigt gleichzeitig die Notwendigkeit, das therapeutische Sistieren der Komplementinhibition als individuelle Einzelfallentscheidung zu behandeln (29) (30). Der lange diagnostische Weg zur final persistierenden Ausschlussdiagnose eines aHUS sowie dessen aussergewöhnlich teure Therapie sind für die beobachtete Zurückhaltung der Krankenkassen verantwortlich. Schlussendlich zeigt die Tatsache, dass die behandelnde Ärzteschaft den nationalen TMA-Expertenbeirat hinzuziehen musste, um das ablehnende Votum des Vertrauensarztes der Krankenversicherung zu überstimmen und die erfolgreiche Therapie mit Eculizumab fortzuführen, wie wichtig ein interdisziplinäres Zusammenspiel bei der Diagnostik und Therapie dieses seltenen Syndroms ist.
Oberarzt
Klinik für Pneumologie
Universitätsspital Zürich
Rämistrasse 100
CH-8091 Zürich
maurice.roeder@usz.ch
Die Autorinnen und Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Gäckler A, Witzke O. Thrombotische Mikroangiopathie. Der Nephrologe. 2021;16(2):113-23.
2. Miller DP, Kaye JA, Shea K, Ziyadeh N, Cali C, Black C, Walker AM. Incidence of Thrombotic Thrombocytopenic Purpura/Hemolytic Uremic Syndrome. Epidemiology. 2004;15(2):208-15.
3. D.R. Terrell LAW. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. Journal of thrombosis and haemostasis. July 2005;3(7).
4. McFarlane PA, Bitzan M, Broome C, Baran D, Garland J, Girard LP, et al. Making the Correct Diagnosis in Thrombotic Microangiopathy: A Narrative Review. Can J Kidney Health Dis. 2021;8:20543581211008707.
5. George JN. Management of immune thrombocytopenia–something old, something new. N Engl J Med. 2010;363(20):1959-61.
6. Holle J, Lange-Sperandio, B., Mache, C., Oh, J., Pape, L., Schaefer, F., Vester, U., Weber, L. T. and Müller, D. Hämolytisch-urämisches Syndrom im Kindes- und Jugendalter. Monatsschrift Kinderheilkunde. 2017;165.
7. Yan K, Desai K, Gullapalli L, Druyts E, Balijepalli C. Epidemiology of Atypical Hemolytic Uremic Syndrome: A Systematic Literature Review. Clin Epidemiol. 2020;12:295-305.
8. Loirat C, Frémeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60.
9. Campistol JM, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35(5):421-47.
10. Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267-79.
11. Ratgeber R. EHEC Erkrankung – Vorkommen. Epidemiologisches Bulletin des Robert Koch Instituts. 2023;Ausgabe 36/2023.
12. Albrecht S, Kamla CE, Schönermarck U, Wassilowsky D. [Thrombotic microangiopathies in surgical intensive care medicine with special consideration of atypical hemolytic uremic syndrome]. Anaesthesiologie. 2023;72(1):3-12.
13. Knoop M, Haller H, Menne J. [Human genetics in atypical hemolytic uremic syndrome-its role in diagnosis and treatment]. Internist (Berl). 2018;59(8):799-804.
14. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: State of the art. Mol Immunol. 2015;67(1):31-42.
15. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-59.
16. Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, Ngo S, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554-62.
17. Thompson GL, Kavanagh D. Diagnosis and treatment of thrombotic microangiopathy. Int J Lab Hematol. 2022;44 Suppl 1(Suppl 1):101-13.
18. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-81.
19. Rondeau E, Scully M, Ariceta G, Barbour T, Cataland S, Heyne N, et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020;97(6):1287-96.
20. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25(11):1256-64.
21. Facharztmagazine R. Zulassung für Komplementinhibitor erweitert. Im Fokus Onkologie. 2021;24(1):58-.
22. Krishnappa V, Gupta M, Elrifai M, Moftakhar B, Ensley MJ, Vachharajani TJ, et al. Atypical Hemolytic Uremic Syndrome: A Meta-Analysis of Case Reports Confirms the Prevalence of Genetic Mutations and the Shift of Treatment Regimens. Therapeutic Apheresis and Dialysis. 2018;22(2):178-88.
23. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to Stop Eculizumab in Complement-Mediated Thrombotic Microangiopathies. Am J Nephrol. 2018;48(2):96-107.
24. Fakhouri F, Fila M, Hummel A, Ribes D, Sellier-Leclerc AL, Ville S, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicenter study. Blood. 2021;137(18):2438-49.
25. Hachem RR, Yusen RD, Chakinala MM, Aloush AA, Patterson GA, Trulock EP. Thrombotic microangiopathy after lung transplantation. Transplantation. 2006;81(1):57-63.
26. Sridharan M, Go RS, Abraham RS, Fervenza FC, Sethi S, Bryant SC, et al. Diagnostic Utility of Complement Serology for Atypical Hemolytic Uremic Syndrome. Mayo Clin Proc. 2018;93(10):1351-62.
27. Mallett A, Hughes P, Szer J, Tuckfield A, Van Eps C, Cambell SB, et al. Atypical haemolytic uraemic syndrome treated with the complement inhibitor eculizumab: the experience of the Australian compassionate access cohort. Internal Medicine Journal. 2015;45(10):1054-65.
28. (ON): APCCINosdO. Pharmacoeconomic Report: Eculizumab (Soliris): Canadian Agency for Drugs and Technologies in Health; . 2020 Oct. ;Appendix 1, Cost Comparison Table.
29. Ardissino G, Testa S, Possenti I, Tel F, Paglialonga F, Salardi S, et al. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis. 2014;64(4):633-7.
30. Merrill SA, Brittingham ZD, Yuan X, Moliterno AR, Sperati CJ, Brodsky RA. Eculizumab cessation in atypical hemolytic uremic syndrome. Blood. 2017;130(3):368-72.
Wir berichten über eine Patientin mit Morbus Basedow, welche wegen typischen Nebenwirkungen auf Carbimazol auf eine Behandlung mit Propylthiouracil umgestellt wurde. Nach 14 Monaten Behandlung mit Propylthiouracil entwickelte sie eine kutane limitierte ANCA-assoziierte Vaskulitis, was eine seltene Nebenwirkung darstellt. Das sofortige Absetzen von Propylthiouracil und eine topische antientzündliche Behandlung führten zur vollständigen Rückbildung der Hautläsionen.
Carbimazol (CMZ) ist die Erstlinientherapie der meisten Patienten mit Morbus Basedow. Propylthiouracil (PTU) wird häufig im ersten Trimenon der Schwangerschaft und bei Nebenwirkungen auf CMZ eingesetzt. Zu den schweren, aber selteneren Nebenwirkungen gehört unter anderem das Auftreten von einer Antineutrophilen zytoplasmatischen Antikörper (ANCA)-assoziierten Vaskulitis. Sie ist klinisch einer primären ANCA-Vaskulitis sehr ähnlich, hat aber meist einen weniger schwerwiegenden Verlauf. Das frühe Absetzen von PTU ist nebst dem Screening auf systemische Manifestationen therapeutisch und prognostisch essenziell. Sowohl für Endokrinolog/-innen, aber auch für die in die Weiterbetreuung involvierten Hausärzt/-innen und Internist/-innen ist das Wissen über seltene Nebenwirkungen von Thyreostatika entscheidend.
Fallvorstellung
Eine 51-jährige Frau präsentierte sich mit Hitzewallungen, Haarausfall, Nervosität, Zittern und Konzentrationsschwäche. Nach der initialen Verdachtsdiagnose eines klimakterischen Syndroms zeigte die hausärztliche Labordiagnostik eine manifeste Hyperthyreose mit einem supprimierten TSH von < 0.001 mlU/l (Referenzbereich 0.35–4,94 mlU/l) und erhöhtem fT4 von 21.4 pmol/l (Referenzbereich 9.01– 19.05 pmol/l) und fT3 von 10.5 pmol/l (Referenzbereich 2.43–6.0 pmol/l). Die ergänzend bestimmten Autoantikörper (TRAK 21.55 U/l, Referenzbereich: < 1.75 U/l) waren diagnostisch für das Vorliegen eines Morbus Basedow (Tab. 1). Klinisch begann der Hausarzt mit 35 mg Carbimazol (CMZ, Neo-Mercazole®) täglich, entschied sich gegen eine zusätzliche symptomatische Behandlung mit einem nicht selektiven Betablocker und wies die Patientin für eine ergänzende Schilddrüsensonographie den endokrinologischen Kollegen zu.
Unter der Medikation entwickelte die Patientin rasch unerwünschte Nebenwirkungen mit einer Stomatitis, Kopfschmerzen sowie Arthralgien, was zu einem Therapiewechsel auf Propylthiouracil (PTU, Propycil® 300 mg/Tag) führte. PTU wurde deutlich besser vertragen und anhand der regelmässigen Laborkontrollen (bei supprimiertem TSH initial anhand des fT4) gelang es, eine baldige Euthyreose und Dosisreduktion zu erreichen. 14 Monate später bemerkte die Patientin an der unteren Extremität neue schmerzhafte, subkutane, hyperpigmentierte Noduli, zunächst am linken Schienbein und später auch an der rechten Wade.
Im Verlauf entwickelten sich die Noduli zu seropurulent gefüllten Bullae, die schliesslich spontan ulzerierten und schmerzhafte Wunden hinterliessen (Abb. 1, 2). Es erfolgte eine dermatologische Zuweisung zur Mitbeurteilung. Aufgrund des klinischen Befundes wurde differenzialdiagnostisch primär an eine infektiöse Pannikulitis mit sporotrichoider Verteilung gedacht. Als Differenzialdiagnosen kamen eine tiefe Mykose, Leishmaniose, Hauttuberkulose oder auch ein Lymphom infrage. Die mikrobiologische Diagnostik einer frisch eröffneten, purulent gefüllten Bulla wies kein Bakterien- oder Pilzwachstum nach und sowohl die Kultur als auch die PCR für Mykobakterien blieb negativ. Histologisch liess sich in der Hautbiopsie eine granulomatöse Dermatitis mit septaler und lobulärer Pannikulitis objektivieren.
Aufgrund dessen und wegen der ausführlich erhobenen Medikamentenanamnese, welche nebst PTU lediglich ein NSAR beinhaltete, das die Patientin wegen den schmerzhaften Hautläsionen einnahm, wurde eine medikamentöse Nebenwirkung näher in Betracht gezogen und eine serologische Testung der Antineutrophilen zytoplasmatischen Antikörper (ANCA) angeordnet. Die Resultate ergaben sowohl einen erhöhten Titer für PR3-ANCA als auch für MPO-ANCA, was in der Zusammenschau der Befunde zur Verdachtsdiagnose einer PTU-induzierten, ANCA-assoziierten Vaskulitis führte. Entsprechend musste nebst einem umgehenden Sistieren der Therapie ein differenziertes Screening hinsichtlich systemischer Manifestationen der Vaskulitis gesucht werden: Untersuchung inkl. Neurostatus, erweiterte Labordiagnostik mit u.a. Blutbild, Entzündungs-, Nieren- und Leberparameter, Urinsediment sowie eine radiologische Diagnostik hinsichtlich einer Lungenbeteiligung. Bei fehlenden Hinweisen auf eine systemische Manifestation konnte schlussendlich von einer kutan limitierten Vaskulitis ausgegangen werden, sodass die Behandlung topisch und nicht systemisch eingeleitet wurde: Clobetasolpropionat (Dermovate®) wurde während zwei Wochen täglich auf die entzündlichen Läsionen aufgetragen, bei gutem Ansprechen dann schrittweise reduziert und auf ein topisches Tacrolimus (Protopic® 0.1 %) gewechselt. Zudem wurde zum Schutz vor einer bakteriellen Superinfektion antiseptische Externa angewendet (Triclosan Softcreme).
Nach einer Behandlungsdauer von 7 Monaten konnte eine deutliche Rückbildung der entzündlichen Hautläsionen, jedoch mit Narbenbildung, beobachtet werden (Abb. 3).
Hinsichtlich des Morbus Basedow wurde der Patientin wegen der Nebenwirkung auf beide gängigen thyreostatischen Medikamente zu einer definitiven Therapie (Radiojod-Behandlung oder Thyroidektomie) geraten, was sie jedoch ablehnte und sich für einen erneuten Versuch mit dem initialen verwendeten CMZ entschied. Dies wurde in der Folge problemlos vertragen. Nach weiteren 10 Monaten Therapie gelang es schlussendlich, die thyreostatische Behandlung nach insgesamt 14 Monaten dokumentierter Euthyreose unter Therapie erfolgreich abzusetzen, und die Patientin blieb euthyreot.
Diskussion
Carbimazol (CMZ, Neo-Mercazole®) und Propylthiouracil (PTU, Propycil®) sind die zwei in der Schweiz verfügbaren Thyreostatika und gehören beide chemisch zu der Gruppe der Thionamide. CMZ ist das Prodrug von Methimazol (MMZ), was beispielsweise in den USA anstatt CMZ verwendet wird. CMZ und MMZ haben im Vergleich zu PTU u.a. wichtige Vorteile hinsichtlich der Halbwertszeit, sodass es einmal täglich eingenommen werden kann, wohingegen PTU auf drei Einnahmen pro Tag verteilt werden muss. Zudem sind unter PTU schwerwiegendere Verläufe von Hepatotoxizität, in äusserst seltenen Fällen gar bis hin zum Leberversagen beschrieben.
Infolgedessen wird es praktisch nur noch bei Unverträglichkeit gegenüber CMZ oder während einer Therapie im 1. Trimenon angewendet. Dort liegt der Vorteil gegenüber CMZ in den weniger schwerwiegenden, embryonalen Fehlbildungen, wobei beide Präparate potenziell teratogen sind. Zu den häufigeren und eher milden Nebenwirkungen beider Präparate zählen Hautreaktionen mit Urtikaria und makulöses Exanthem (4–6 %), Arthralgien (1–5 %), gastrointestinale Beschwerden mit Dyspepsie und Nausea (1–5 %) (1). Schwere Nebenwirkungen unter thyreostatischer Therapie sind zum Glück selten, beinhalten aber das Auftreten von Pankreatitiden (CMZ), Hepatotoxizität, eine ANCA-assoziierte Vaskulitis (PTU) oder die den meisten Klinikern bekannte Agranulozytose (CMZ oder PTU) (Tab. 2) (1). Die Prävalenz letzterer liegt gemäss Literatur bei rund 0.1–0.5 %, und es sind Kreuzreaktionen beschrieben. Thyreostatika-induzierte Agranulozytosen treten in den allermeisten Fällen innerhalb der ersten rund drei Monate der Therapie auf, sodass auch von einer Dosisabhängigkeit ausgegangen wird (zumindest für CMZ).
Interessanterweise konnten genetische Analysen das Risiko einer Agranulozytose mit gewissen HLA-Typen in Verbindung bringen, was aber aufgrund der Seltenheit der Nebenwirkung nicht im klinischen Alltag vor einem Therapiebeginn untersucht wird (3). Umso mehr müssen Betroffene vor Beginn der Therapie durch die Behandlungsperson eingehend über diese Nebenwirkung aufgeklärt werden und wissen, dass sie im Falle einer akuten febrilen Erkrankung, häufig mit Pharyngitis begleitet, das Präparat absetzen und sich zeitnah für eine Blutbild-kontrolle melden müssen.
Von einer generellen laborchemischen Überwachung der Patientinnen und Patienten unter Therapie raten wichtige internationale Guidelines ab (2, 4). Erhöhte Transaminasen und insbesondere eine erhöhte Gamma-GT ist ein relativ häufiger Befund einer Hyperthyreose bei Morbus Basedow und daher von einer medikamentösen Nebenwirkung durch Thyreostatika abzugrenzen. Sie normalisieren sich unter der Behandlung und dem Erreichen einer Euthyreose. In diesen Fällen sollten die Leberparameter zusammen mit der Schilddrüsenfunktion initial regelmässig kontrolliert werden, um eine Abgrenzung hinsichtlich einer Hepatotoxizität durch die Therapie zu ermöglichen (6).
Die in diesem Fall beschriebene ANCA-assoziierte Vaskulitis stellt eine überaus seltene Nebenwirkung einer thyreostatischen Therapie mit Thionamiden dar und ist fast ausschliesslich für PTU beschrieben. In der Regel kam es – wie in unserem Fall – deutlich verzögert (mehrere Monate bis sogar Jahre nach Behandlungsbeginn) zum Auftreten, sodass der Zusammenhang bei der initialen Beurteilung primär unterschätzt werden kann.
Die Diagnosestellung erfolgt bei passender Klinik und gegebenenfalls unterstützendem Befund in der Histologie einer biopsierten Läsion mittels Nachweis von erhöhten ANCA-Titern.
Anders als bei anderen ANCA-assoziierten Vaskulitiden scheint es durch PTU zu einer unspezifischen Aktivierung mehrerer ANCA zu kommen, sodass sowohl der Nachweis von Antikörpern gegen Myoloperoxidase und Proteinase-3 beschrieben ist. Ein generelles laborchemisches Screening während einer Therapie mit PTU wird nicht empfohlen, da erhöhte ANCA-Titer bei bis zu 60 % asymptomatischer Patienten im Verlauf gefunden wurden und damit der positive prädiktive Wert sehr limitiert ist (5). Entscheidend bei der Diagnosestellung ist die Diagnostik hinsichtlich systemischer Manifestationen der Vaskulitis. Bei Hinweisen auf beispielsweise einer pulmonalen oder renalen Beteiligung wurde in den in der Literatur beschriebenen Fällen nebst dem Absetzen des auslösenden Agens stets mit einer hoch dosierten, systemischen Glukokortikoidtherapie und im Verlauf allenfalls anderen Immunsuppressiva wie Cyclophosphamid, Azathioprin, Mycophenolat mofetil behandelt. Bei einer kutan limitierten Erkrankung kann wie im geschilderten Fall eine topische antientzündliche Behandlung ausreichen.
Hinsichtlich der thyreostatischen Therapie bestätigt dieser Fall die verfügbare Datenlage, dass ein Wechsel auf CMZ möglich ist und keine Kreuzreaktionen im Sinne eines persistierenden Triggers für die Vaskulitis resultiert.
Schlussfolgerung
Zusammenfassend illustriert dieser Fall eine schwerwiegende, wenngleich seltene Nebenwirkung von PTU in Form einer ANCA-assoziierten Vaskulitis der Haut. Im Vergleich zu primären ANCA-assoziierten Vaskulitiden tritt die PTU-induzierte Vaskulitis häufiger bei jungen Frauen auf und hat bei früher Diagnosestellung in der Regel eine bessere Prognose. Da das Auftreten meist mehrere Monate nach Beginn der Behandlung auftritt, muss das Auftreten kutaner Veränderungen jederzeit an diese Nebenwirkung denken lassen und bei Verdacht entsprechend zeitnahe Abklärungen in die Wege geleitet werden. Ein laborchemisches Screening ist einerseits wegen der Seltenheit der Nebenwirkung und andererseits wegen der geringen Sensitivität der ANCA-Antikörper für die Entwicklung einer klinischen Vaskulitis unangebracht. Bei Diagnosestellung bedarf es eines Screenings hinsichtlich anderer Organbeteiligungen, wobei insbesondere auf eine Affektion der Lunge, Niere und des Nervensystems geachtet werden muss. Je nach Resultaten kann die Initialbehandlung topisch antientzündlich erfolgen oder muss mit systemischen Immunsuppressiva angegangen werden. Das frühzeitige Sistieren der PTU-Behandlung ist in jedem Falle zentral. Hinsichtlich der Hyperthyreose kann ein Therapiewechsel auf CMZ ohne Gefahr von Kreuzreaktionen erfolgen, wobei aber auch über eine definitive Behandlung mittels Thyroidektomie oder Radiojod-Behandlung individuell beraten werden muss.
Fachassistenzarzt
Klinik für Endokrinologie, Diabetologie,
Osteologie und Stoffwechselerkrankungen
Kantonsspital St. Gallen
markus.koster@kssg.ch
Dr. med. Sven Lieberherr
Klinik für Dermatologie, Venerologie und Allergologie,
Kantonsspital St. Gallen,
St. Gallen
Prof. Dr. med. Dr. sc. nat. Antonio Cozzio
Kantonsspital St. Gallen
Rorschacher Strasse 95
Haus 20
9007 St. Gallen
antonio.cozzio@kssg.ch
PD Dr. med. Stefan Bilz
Klinik für Endokrinologie, Diabetologie,
Osteologie und Stoffwechselerkrankungen
HOCH Kantonsspital St. Gallen
Rorschacher Strasse 95
9007 St. Gallen
stefan.bilz@h-och.ch
Die Autorin und die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Cooper DS. Antithyroid drugs. N Engl J Med. 2005;352(9):905-17.
2. Kahaly GJ, Bartalena L, Hegedus L, Leenhardt L, Poppe K, Pearce SH. 2018 European Thyroid Association Guideline for the Management of Graves’ Hyperthyroidism. Eur Thyroid J. 2018;7(4):167-86.
3. Chen PL, Shih SR, Wang PW, Lin YC, Chu CC, Lin JH, et al. Genetic determinants of antithyroid drug-induced agranulocytosis by human leukocyte antigen genotyping and genome-wide association study. Nat Commun. 2015;6:7633.wwww
4. Ross DS, Burch HB, Cooper DS, Greenlee MC, Laurberg P, Maia AL, et al. 2016 American Thyroid Association Guidelines for Diagnosis and Management of Hyperthyroidism and Other Causes of Thyrotoxicosis. Thyroid. 2016;26(10):1343-421.
5. Balavoine AS, Glinoer D, Dubucquoi S, Wemeau JL. Antineutrophil Cytoplasmic Antibody-Positive Small-Vessel Vasculitis Associated with Antithyroid Drug Therapy: How Significant Is the Clinical Problem? Thyroid. 2015;25(12):1273-81.
6. Hsieh A, Adelstein S, McLennan SV, Williams PF, Chua EL, Twigg SM. Liver enzyme profile and rogression in association with thyroid autoimmunity in Graves’ disease. Endocrinol. Diab Metab 2019;2:e00086