Fallbericht
Anamnese und Befunde
Eine 58-jährige Patientin stellte sich notfallmässig mit Schwindel und Nausea sowie einem Ganzkörpererythem und pektanginösen Beschwerden nach dem Verzehr eines Thunfischsteaks in einem Restaurant vor. Bis auf eine Kälteurtikaria waren keine Vorerkrankungen bekannt. Initial bemerkte die Patientin direkt nach Einnahme einen metallisch-scharfen Geschmack auf der Zunge. Die Symptome entwickelten sich in voller Ausprägung, nachdem die Patientin zu Hause ankam, sodass sie drei Stunden nach dem Verzehr des Thunfischs die Ambulanz verständigte. Die Sanitäter vermuteten zuerst eine allergische Reaktion und verabreichten Flüssigkeit, intramuskuläres Adrenalin sowie Steroide und Clemastin.
Kurze Anamnese mit Betonung des jetzigen Leidens
Im Spital zeigte sich die Patientin initial hypotensiv (91/47 mmHg), sodass die intravenöse Flüssigkeitszufuhr fortgesetzt wurde. In der körperlichen Untersuchung ergab sich ein diffuses Erythem am ganzen Körper, aber ein vesikuläres Atemgeräusch ohne Stridor. Die kardiale Untersuchung erbrachte keinen pathologischen Befund. Auf der Notfallstation beschrieb die Patientin keine respiratorischen Symptome und benötigte keinen Sauerstoff.
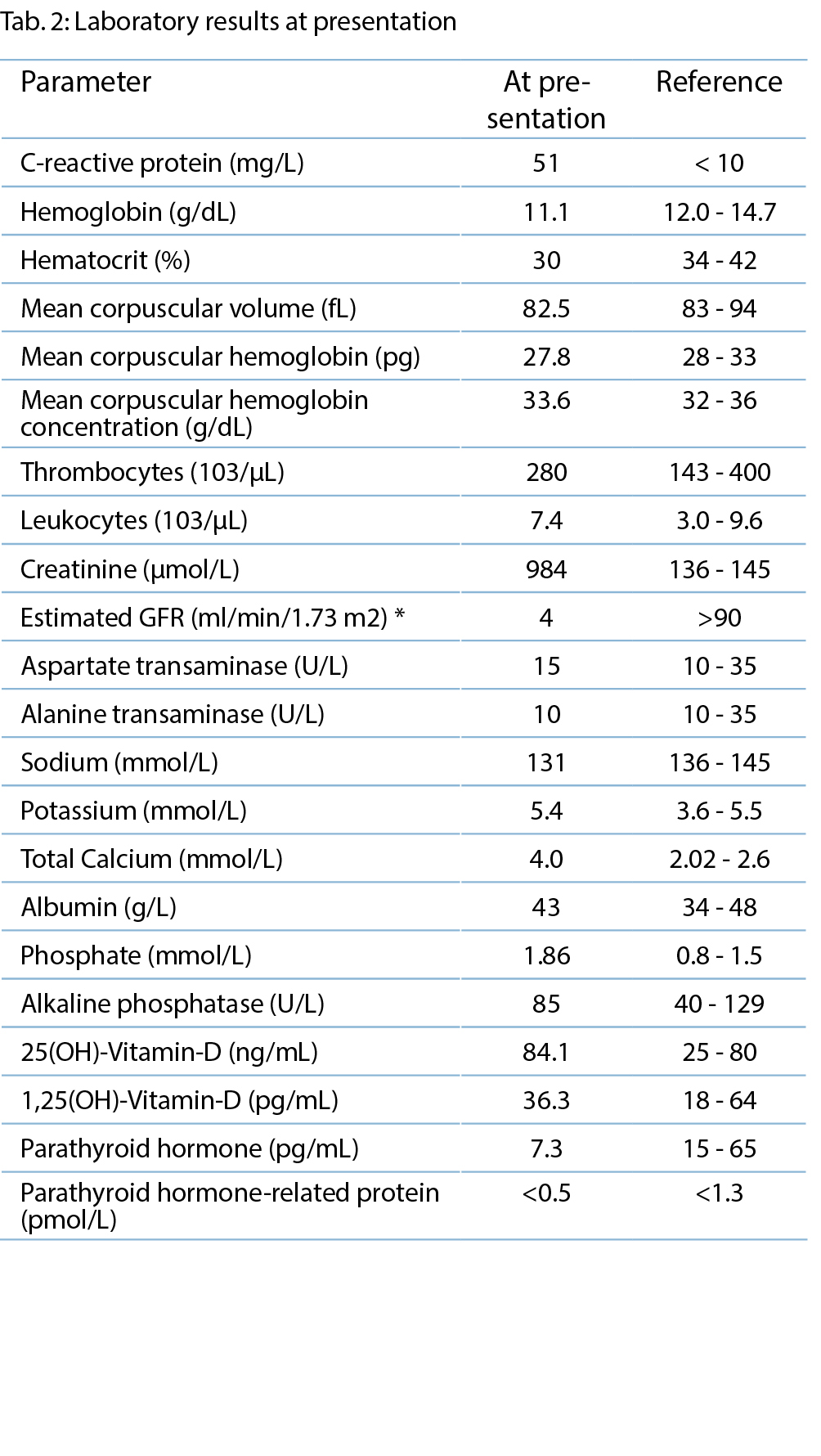
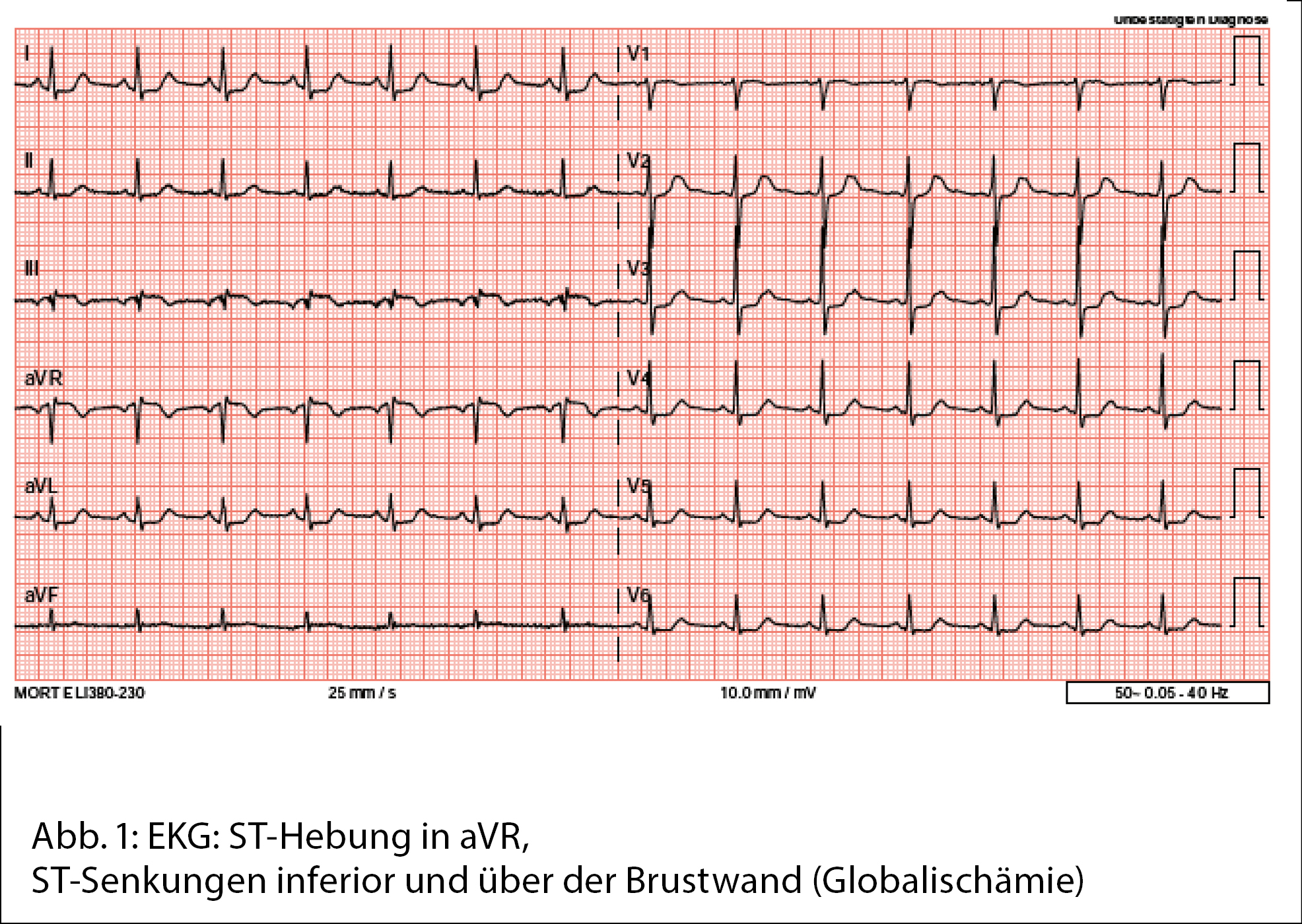
Laborchemisch konnte eine mässige Leukozytose sowie ein erhöhtes Kreatinin nachgewiesen werden. Troponin- T-Analyse und Blutalkoholspiegel ergaben Normalbefunde. Die Tryptase-Werte bei der Aufnahme waren normal. Nach einer zweiten intramuskulären Adrenalingabe zeigte die Patientin normotone Blutdruckwerte, und das Erythem war vollständig regredient. Im initialen Elektrokardiogramm (EKG) zeigten sich diffuse minimale ST-Senkungen (Abb. 1), welche in der Verlaufskontrolle nach einer Stunde viel deutlicher wurden und sich in einem Hauptstamm-EKG-Muster präsentierten (Abb. 2).
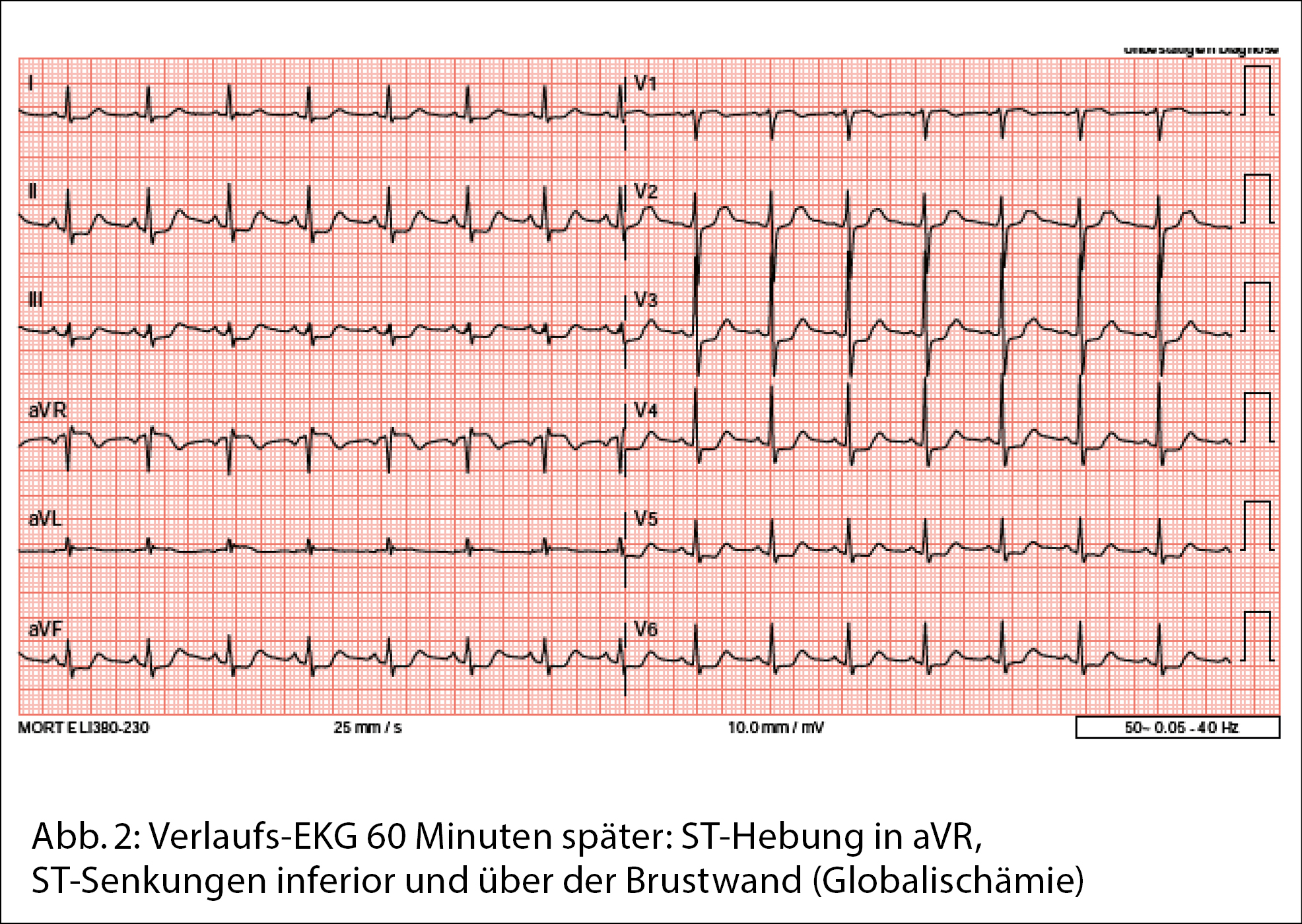
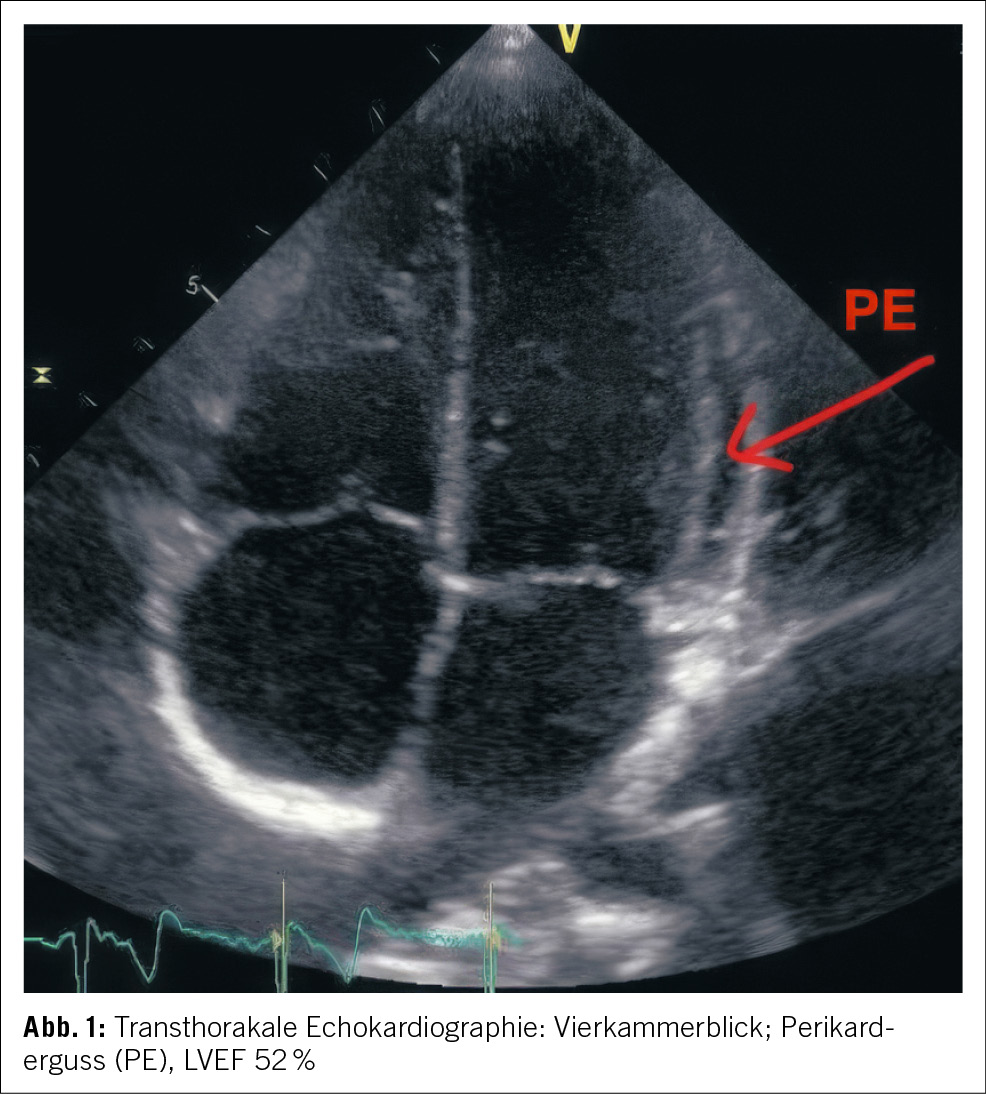
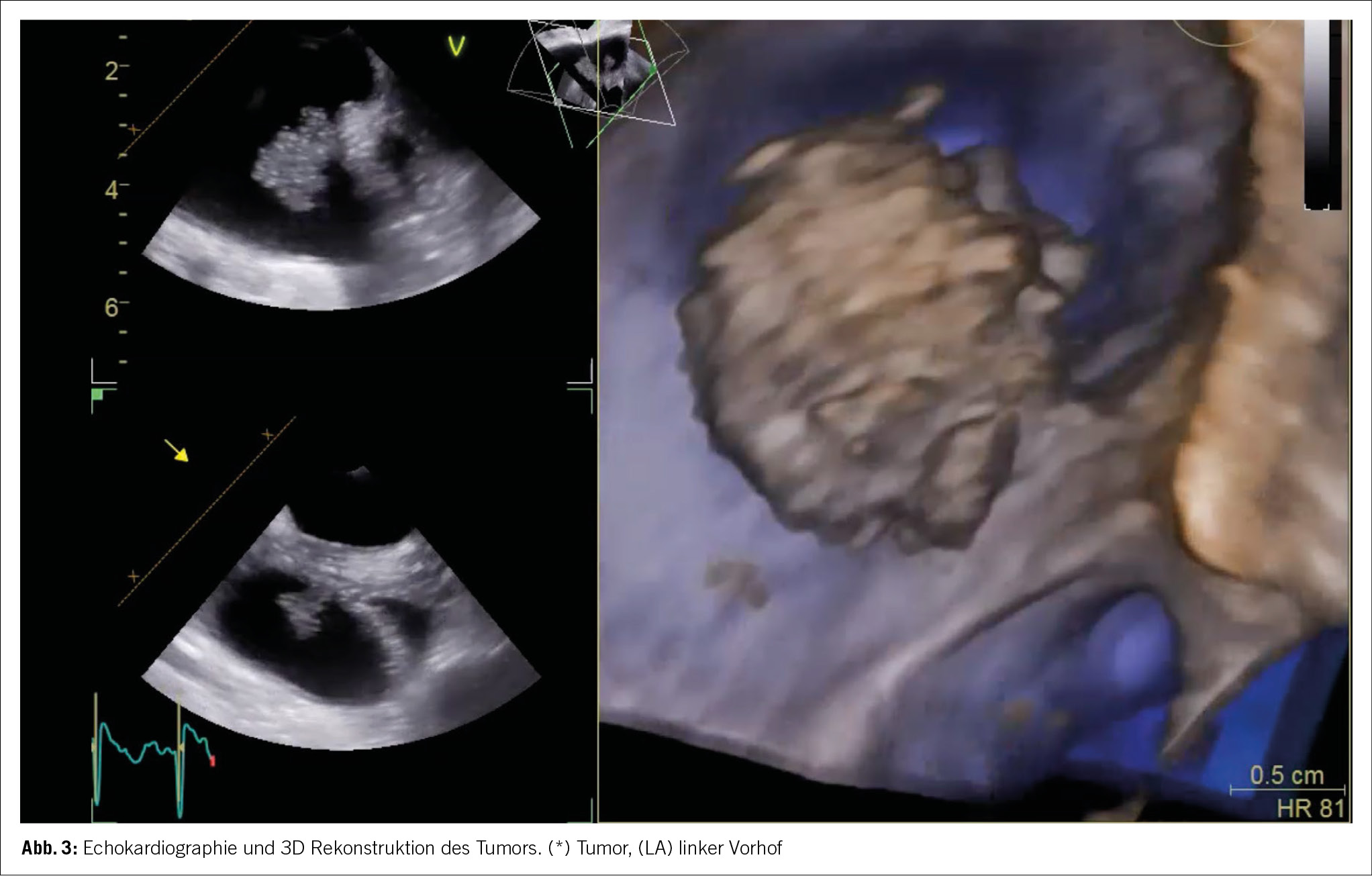
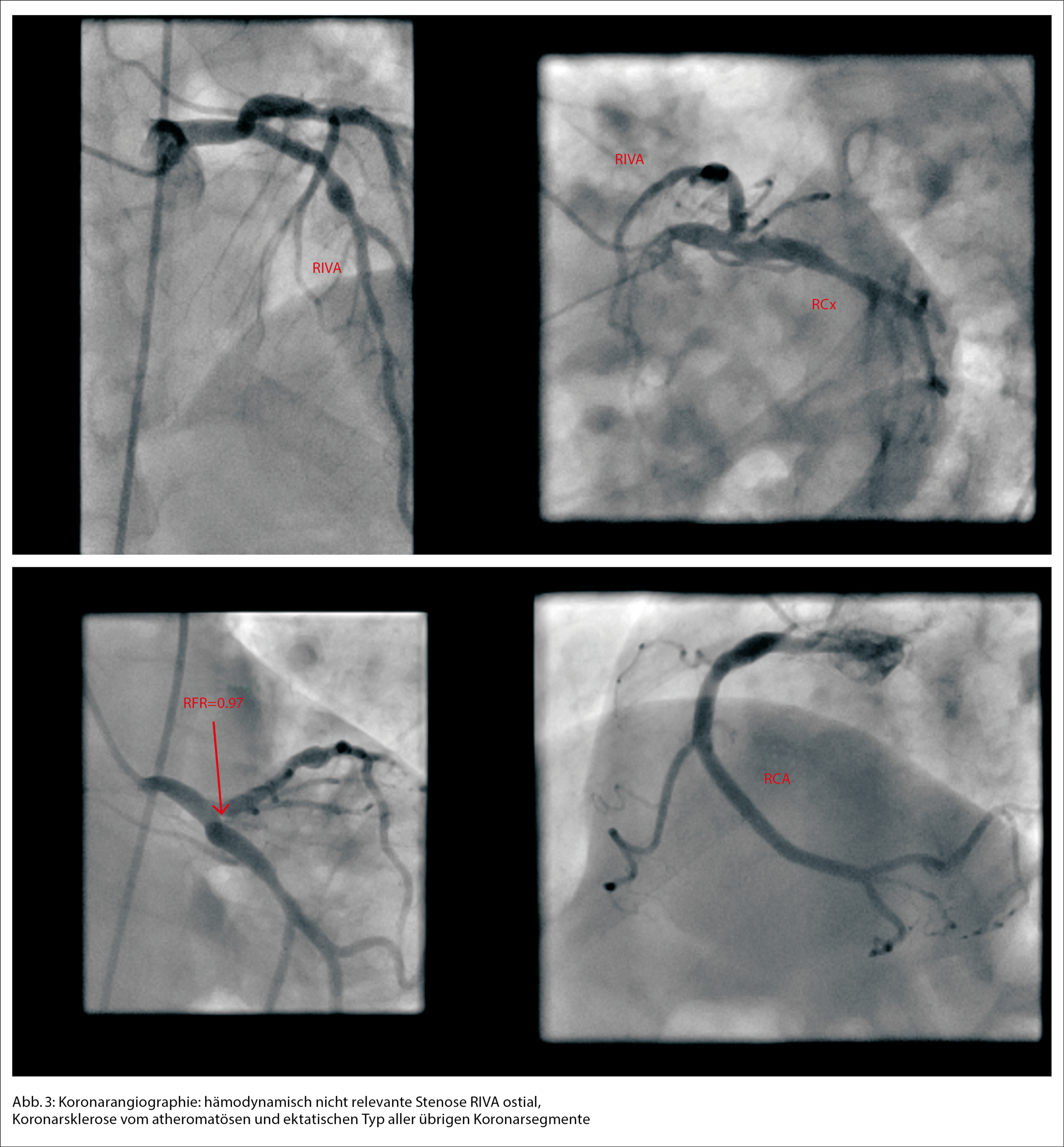
Die Patientin wurde zur weiteren Überwachung auf unsere Intermediate Care Station aufgenommen. In den folgenden Laborkontrollen stieg das kardiale Troponin T deutlich an, was uns veranlasste, unser kardiologisches Team hinzuziehen, da wir ein akutes Koronarsyndrom befürchteten. Die Echokardiographie ergab eine normale Ejektionsfraktion und keinen Hinweis auf Wandbewegungsstörungen. In der Koronarangiographie imponierten die Koronargefässe mit lediglich einer moderaten Koronarsklerose ohne Stenosierung (Abb. 3). In Verbindung mit dem Hauptstamm-EKG wurde ein Koronarspasmus durch die Adrenalingaben postuliert, es zeigten sich in der Koronarangiographie jedoch keine Hinweise für eine höhere Spasmusneigung. Während des Krankenhausaufenthalts zeigten die Folge-EKGs eine Verbesserung der ST-Streckenveränderungen und schliesslich eine Normalisierung.
Der Troponin-T-Wert erreichte einen Spitzenwert
von 192 ng/ml (0–26 ng/ml) mit anschliessender Normalisierung. Die Tryptase zeigte sich auch in der wiederholten Kontrolle am zweiten Tag des Spitalaufenthalts
normwertig.
Alle Symptome klangen innerhalb von 12 Stunden nach der notfallmässigen Vorstellung ab, und die Patientin konnte nach Hause entlassen werden.
Differenzialdiagnostische Überlegungen
Die systemische anaphylaktoide Reaktion mit Beginn Stunden nach der Einnahme des kontaminierten Fisches ist sehr suggestiv für eine Histaminintoxikation. Das negative Ergebnis der Tryptase-Analyse passt auch zu einer Histaminintoxikation.
In dem hier beschriebenen Fall wurde ein Typ 1 Kounis-Syndrom vermutet, da die Symptome nach Behandlung der anaphylaktoiden Reaktion verschwanden und in der angiographischen Untersuchung keine kritische Koronarerkrankung gefunden wurde. Als Differenzialdiagnosen der systemischen anaphylaktoiden Reaktion mit Beginn Stunden nach der Einnahme des Fisches kamen eine klassische Anaphylaxie mit dem pathogenetischen Mechanismus der Mastzelldegranulation oder eine direkte Histaminintoxikation durch die Einnahme des möglicherweise kontaminierten Fisches infrage. Das repetitiv negative Ergebnis der Tryptase-Analyse passte zu einer Histaminintoxikation.
Bei gleichzeitigem Vorhandensein eines akuten Koronarsyndroms ohne Nachweis einer kritischen Stenose in der Koronarangiographie war ein Typ 1 Kounis-Syndrom möglich, wobei das Kounis-Syndrom bislang nur im Rahmen von klassischen Anaphylaxien mit Mastzelldegranulation und weniger im Rahmen von Histaminintoxikationen beschrieben wurde.
Da in der Schweiz jedoch vermehrt Fisch konsumiert wird, wird die Inzidenz des Kounis-Syndroms im Rahmen von Histaminintoxikationen zunehmen.
Ein wie von den Kardiologen vermuteter, durch Adrenalin verursachter Vasospasmus wurde differenzialdiagnostisch in Betracht gezogen. Dieser würde jedoch die Persistenz der pektanginösen Beschwerden und das Fehlen einer Spasmusneigung in der Koronarangiographie nicht erklären.
Weitere Abklärungsschritte
Eine allergologische Abklärung wurde empfohlen. Hier fand sich keine Sensibilisierung auf Fisch oder Soja. Da sich die Patientin gegen Ende der Konsultation auch nicht sicher war, ob allenfalls Baumnüsse im Gericht waren, wurde auch eine serologische Bestimmung diesbezüglich durchgeführt, jedoch konnten wir auch keine Sensibilisierung auf Walnuss feststellen.
Diagnose
Scombroid-Vergiftung mit Typ 1 Kounis-Syndrom durch die Histaminintoxikation
Kommentar
Scombroid-Vergiftungen können nach dem Verzehr von nicht fachgerecht gelagertem Fisch vorkommen. Fischfleisch enthält Histidin. Wenn der Fisch durch gramnegative Bakterien kontaminiert ist, spaltet das bakterielle Enzym Histidin-Decarboxylase das Histidin zu Histamin. Das exogene eingenommene Histamin führt zu einer allergieähnlichen Reaktion, wie es ebenfalls bei einer Allergie-induzierten Histaminantwort durch die Degranulation von Mastzellen kommen kann (4).
Diese Reaktionen sind jedoch nicht IgE- oder gar Allergen-vermittelt, sondern werden direkt durch das im Fisch enthaltene Histamin ausgelöst.
Tox Info Suisse registriert jedes Jahr circa 10 bestätigte Fälle dieser Art von Lebensmittelvergiftung. Die Symptome einer Scombroid-Vergiftung sind sehr ähnlich wie die einer klassischen Anaphylaxie und können leicht verwechselt werden. Am häufigsten treten Scombroid-Vergiftungen nach Fischkonsum auf. Die betroffenen Fische sind Makrelen (Scombroide), Thunfische (Thunis spp.), Bonitos (Sarda spp.), Makrelenhechte (Scombroidecidea), Stachelmakrelen (Caragidae), Heringe und Sardinen (Clupeidae), Anchovis und Sardellen (Engraulidae) sowie Blaufische (Pomatoidae). Insbesondere Makrelen gehören zu einer Gruppe von Fischen mit hohem Histamingehalt.
Das Kounis-Syndrom wird klinisch als Kombination eines akuten Koronarsyndroms mit einer Histamin-induzierten anaphylaktischen oder anaphylaktoiden Reaktion definiert, wobei es über eine Mastzelldegranulation zu einer Histaminliberation gekommen ist (6). Die Diagnose des Kounis-Syndroms ist nicht trivial und erfordert viel Erfahrung. Die klinischen Symptome einer allergischen oder anaphylaktoiden Reaktion sowie die laborchemischen und elektrokardiographischen Befunde einer akuten kardialen Ischämie sollten den Verdacht auf ein Kounis-Syndrom lenken.
Es sind drei Formen des Kounis-Syndroms bekannt (3):
– Typ 1 tritt bei Patienten auf, die keinen Hinweis auf eine koronare Herzerkrankung haben. Hierbei ist pathophysiologisch die endotheliale Dysfunktion mit konsekutivem Vasospasmus Grund für die Symptomatik.
– Typ 2 tritt bei Patienten mit nicht kritischen arteriosklerotischen Veränderungen auf. Die Symptomatik ist dabei durch Koronarspasmen mit Einreissen von atherosklerotischen Plaques und konsekutiver Thrombosierung der Wandverletzungen zu erklären.
– Typ 3 bezieht sich auf allergische Reaktionen gegen Komponenten der koronaren Herzerkrankung mit konsekutiver in-Stent-Thrombose.
Sekundäre, durch allergische Reaktionen verursachte, akute Koronarsyndrome sind mit einer signifikant erhöhten Morbidität und Mortalität verbunden und benötigen daher eine intensivmedizinische Überwachung (3). Das klinische Management des Kounis-Syndroms ist eine Herausforderung, da die kardialen sowie die allergischen oder (wie in diesem Fall) anaphylaktoiden Symptome gleichzeitig behandelt werden müssen. Bei Patienten mit einer Typ-1-Variante kann allein die Therapie der allergischen oder anaphylaktoiden Reaktion die Symptome beseitigen.
Adrenalin ist die Standardbehandlung der Anaphylaxie, kann jedoch eine myokardiale Ischämie aggravieren und einen Koronarspasmus verschlechtern. Es sollte daher unter überwachten Bedingungen verabreicht werden. Unproblematisch sind intravenöse Steroide sowie H1- und H2-wirksame Antihistaminika. Die Behandlung ischämischer Schmerzen beim Kounis-Syndrom weicht ebenfalls von den Standardbehandlungsprotokollen ab, da sich Opioide wie Morphin auf die Mastzelldegranulation auswirken und somit die allergische Reaktion aggravieren können. Fentanyl als Opioid mit der geringsten Auswirkung auf die Mastzelldegranulation ist daher die empfohlene Wahl der Therapie bei diesen Patienten (3).
Die Messung der Serumtryptase als Marker der Mastzellaktivierung ist nicht immer diagnostisch zuverlässig. Da sie nur eine sehr kurze Halbwertszeit von circa 90 Minuten hat, sollte die erste Probe eine halbe Stunde nach Beginn der initialen Symptome entnommen werden und weitere Proben nach 30 Minuten respektive nach 2 Stunden. Ähnlich kurzwirksam ist Histamin mit einer Halbwertszeit von ca. 8 Minuten, was die laborchemische Bestimmung ebenfalls massiv erschwert. Zudem ist sie stark beeinflusst von Analgetika-Einnahmen, insbesondere Morphin (3).
Anamnestisch berichtete die Patientin bei Eintritt, bis auf eine Kälteurtikaria ohne Dauertherapie keine Krankheiten zu haben. In der Vergangenheit wurde sie allerdings bei wiederholt aufgetretenen, unklaren Thoraxschmerzen kardiologisch abgeklärt, was keine richtungsweisenden Befunde ergab. In der erweiterten Anamnese auf der Abteilung gab die Patientin an, dass die thorakalen Beschwerden bei Kälte und zum Beginn körperlicher Belastung (zum Beispiel Jogging) stärker seien, im Verlauf einer Trainingseinheit aber vollständig verschwänden.
Eine mögliche Assoziation zwischen der Histaminfreisetzung im Rahmen einer Kälteurtikaria und dem Koronarspasmus beim Typ 1 Kounis-Syndroms wurde schon in einzelnen seltenen Fällen in der Literatur beschrieben (5).
Unserer Meinung nach ist in unserem Fall eine Spasmusneigung der Koronararterien gegen Histamin möglich und kann durch einen Provokationstest in der Koronarangiographie nicht ausgeschlossen werden, da dieser unter einer vollen Antihistaminikatherapie erfolgte.
Auch wenn eine allergologische Abklärung im Rahmen des Work-up einer Scombroid-Vergiftung normalerweise nicht indiziert ist, haben wir in diesem Fall diese Möglichkeit mit der Patientin diskutiert und daher eine immuno-allergologische Standortbestimmung durchgeführt, welche keine Hinweise auf Fischsensibilisierung zeigte.
Streng genommen beschreiben wir hier eine neue Entität des Kounis-Syndroms Typ 1, wobei das Koronarspasmus-induzierende Histamin nicht von einer Mastzelldegranulation stammte, sondern direkt von exogen zugeführt wurde. Der Zusammenhang zwischen Scombroid-Intoxikationen und dem Auftreten eines solchen Kounis-Syndroms ist in der Literatur bereits dokumentiert, vor allem durch Fälle aus Mittelmeerländern (2).
Mit den veränderten Essgewohnheiten der Schweizer Bevölkerung (mehr Verzehr von Fisch) werden solche Fälle in Zukunft zunehmen.
Historie:
Manuskript eingereicht: 28.11.2023
Angenommen nach Revision: 30.04.2024
Ärztlicher Leiter
Intensivstation und Intermediate Care Station
Kantonsspital Olten
Baslerstr 150
4600 Olten
michael.studhalter@spital.so.ch
Es bestehen keine Interessenkonflikte.
1. www.toxinfo.ch
2. Cesare de Gregorio, Giuseppe Ferrazzo, Ioanna Koniari & Nicholas G. Kounis (2022) Acute coronary syndrome from scombroid poisoning: a narrative review of case reports, Clinical Toxicology, 60:1, 1–9.
3. Kounis, Nicholas G. “Kounis syndrome: an update on epidemiology, pathogenesis, diagnosis and therapeutic management” Clinical Chemistry and Laboratory Medicine (CCLM), vol. 54, no. 10, 2016, pp. 1545–1559.
4. Ridolo E, Martignago I, Senna G, Ricci G. Scombroid syndrome: it seems to be fish allergy but… it isn’t. Curr Opin Allergy Clin Immunol. 2016 Oct;16(5):516–21.
5. Mazarakis A, Bardousis K, Almpanis G, Mazaraki I, Markou S, Kounis NG. Kounis syndrome following cold urticaria: the swimmer‘s death. Int J Cardiol. 2014 Sep 20;176(2):e52-3.
6. Khan K, Szalai G, Anjum H, Dimtri F, Yamamura D, Surani S.: Bee Attack or Heart Attack: Kounis Syndrome. Cureus. 2021; 13(4): e 14740. Epub 2021 Apr 28.