Das Long-QT-Syndrom (LQTS) ist eine angeborene Ionenkanalerkrankung, die eine verlängerte ventrikuläre Repolarisation verursacht und sich im Oberflächen EKG mit einer verlängerten QT-Zeit präsentiert. Diese Erkrankung prädisponiert für lebensbedrohliche ventrikuläre Arrhythmie sowie den plötzlichen Herztod. Das LQTS ohne entsprechende Therapie stellt während der Schwangerschaft und in der postnatalen Phase aufgrund der mit der Gestation verbundenen physiologischen Veränderungen ein zusätzlich erhöhtes Risiko für einen plötzlichen Herztod dar. Wir präsentieren einen Fallbericht einer 30-jährigen schwangeren Frau mit bekanntem Long-QT-Syndrom Typ 2 (LQT2) und dem konsekutiven kardiologischen Management.
Einführung
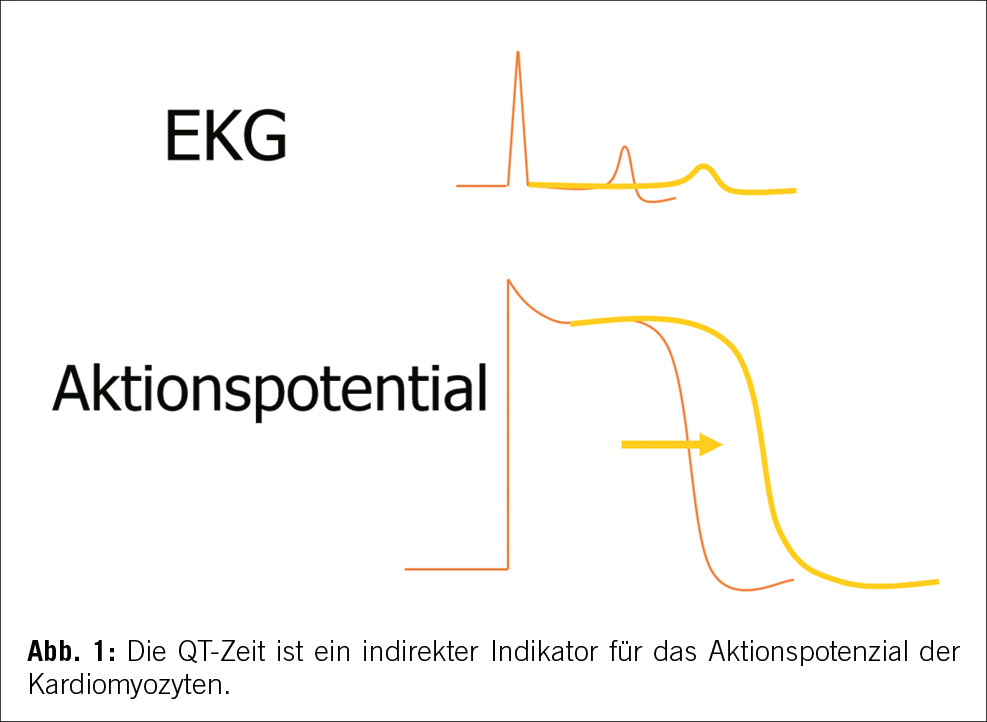
Das angeborene Long-QT-Syndrom ist eine genetische Erkrankung der Ionenkanäle (Kanalopathie), charakterisiert durch eine prolongierte ventrikuläre Repolarisation (QT- Zeit Verlängerung im Ruhe EKG, siehe Abbildung 1). Bei adrenerger Aktivierung kann diese verlängerte Repolarisation – insbesondere bei LQTS 1 und 2 – zu polymorphen ventrikulären Tachykardien bekannt als ,,Torsade de pointes” mit konsekutiven Synkopen bis hin zu einem plötzlichen Herztod führen können [1,2,3].
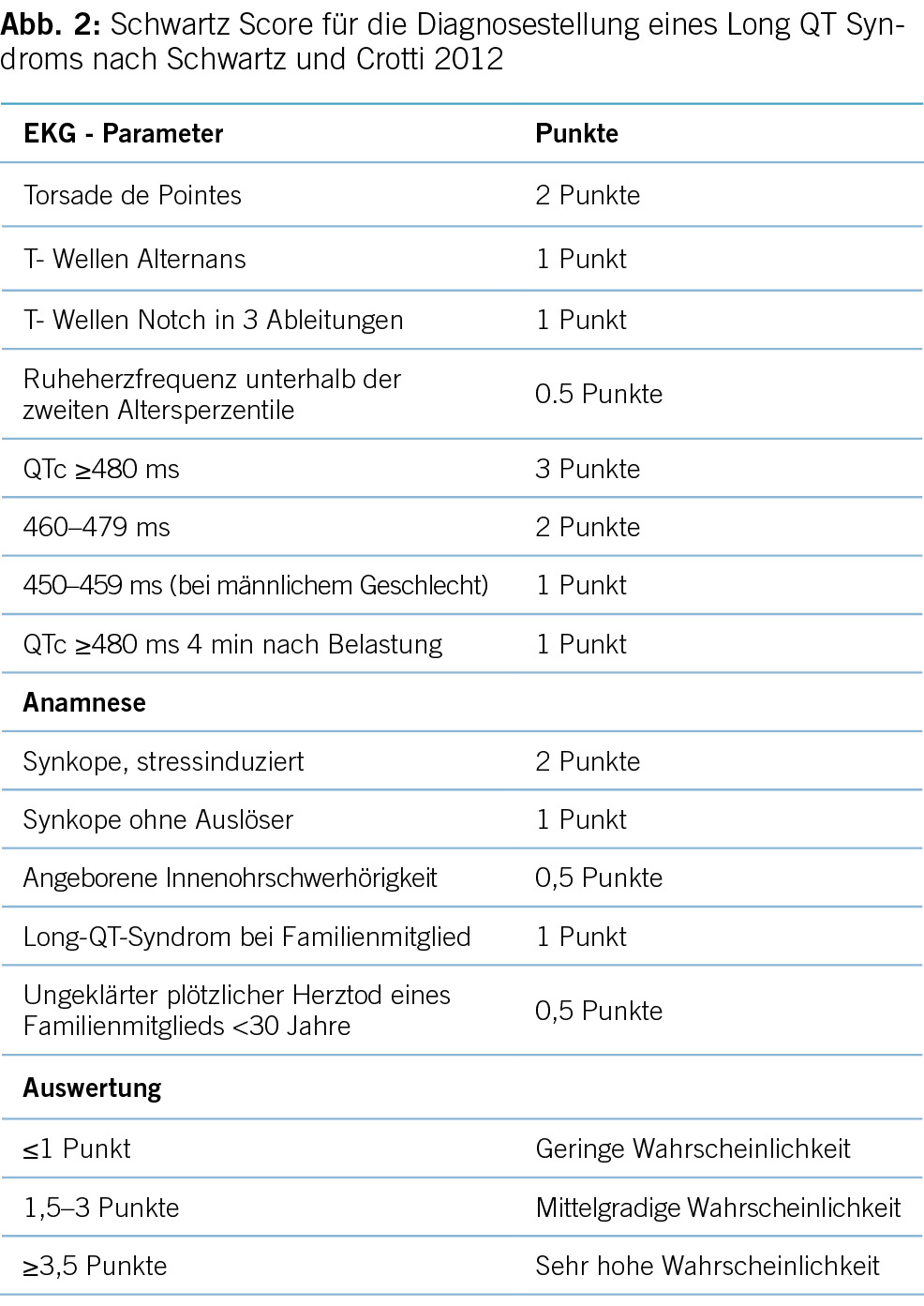
Die korrekte Diagnose des Long-QT-Syndroms (LQTS) basiert auf der herzfrequenzkorrigierten QT-Zeit (QTc), einer Reihe anderer elektrokardiographischer Parameter sowie Informationen aus der Anamnese und Familienanamnese und ggf. der genetischen Untersuchung. Diagnosekriterien nach Schwartz et al. sind state-of-the-art. Wichtig ist zudem das Fehlen von strukturellen Herzerkrankungen, QT-Zeit verlängernden Medikamenten (siehe Abbildung 2) und anderen prädisponierenden Faktoren wie Hypokaliämie [4,5,6].
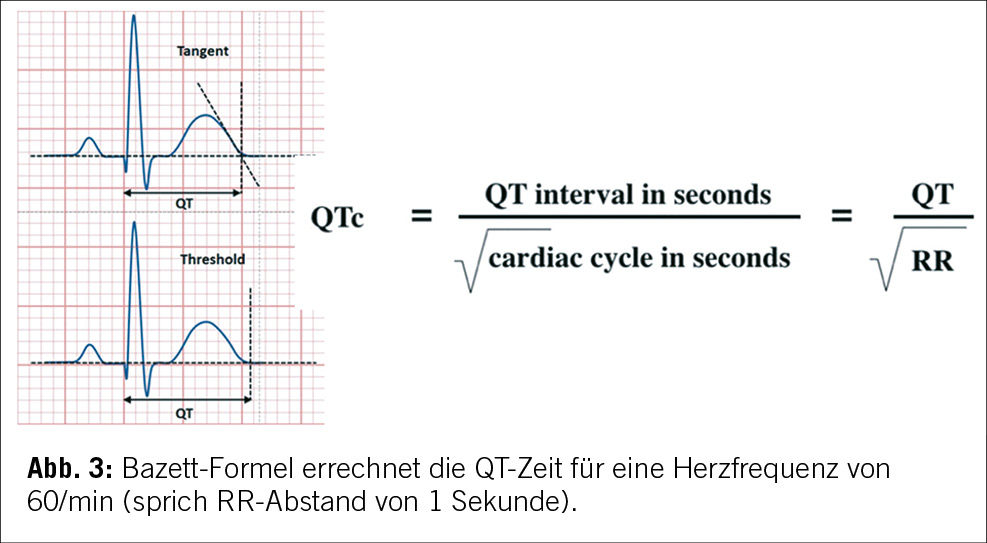
Es gibt verschiedene Methoden zur Messung der QT-Zeit, die zu unterschiedlichen Grenzwerten führen, in der Literatur am besten validiert ist die Bazett- Formel (siehe Abbildung 3). Darüber hinaus sollte, obwohl die U-Welle bei LQTS-Patienten ebenfalls abnorm sein kann, die U-Welle nicht in die QT-Zeit Messung einbezogen werden [6].
Zusätzlich sollten die Kriterien nach Priori et al. berücksichtigt werden, welche bei zutreffen von einer oder mehreren der folgenden Kriterien von einem LQTS ausgehen [7]:
- Bei einem Vorliegen von einem LQTS-Risikowertes ≥3,5 (siehe Abbildung 2) in Abwesenheit einer sekundären Ursache für QT-Verlängerung und/oder
- bei Vorliegen einer zweifelsfrei pathogenen Mutation in einem der LQTS-Gene oder
- bei Vorliegen eines, unter Verwendung von Bazett- Formel korrigierten, QT-Intervalls (QTc) von ≥500 ms in wiederholten 12-Kanal-Elektrokardiogrammen (EKG) und in Abwesenheit einer sekundären Ursache für QT- Zeit Verlängerung.
- Zudem kann LQTS diagnostiziert werden, wenn in wiederholten 12-Kanal-EKGs bei einem Patienten mit ungeklärter Synkope in Abwesenheit einer sekundären Ursache für QT-Verlängerung und in Abwesenheit einer pathogenen Mutation eine QTc- Zeit zwischen 480–499 ms vorliegt.
Das durchschnittliche Alter der ersten Symptome bei LQTS beträgt 14 Jahre. Das jährliche Risiko für einen plötzlichen Herztod (SCD) bei asymptomatischen, unbehandelten LQTS-Patienten wurde auf weniger als 0,5% beziffert, während das jährliche Risiko für einen plötzlichen Herztod auf etwa 5% bei Patienten mit bereits erlittenen Synkopen in der Vorgeschichte ansteigt [6]. Bei symptomatischen Index Patienten beträgt die unbehandelte 10-Jahres-Sterblichkeitsrate sogar um 50% [8]. Es wurden bis dato 11 Gene mit LQTS in Verbindung gebracht, die Prävalenz liegt bei 1:2500 [4,7,10]. Die häufigsten Gene sind diejenigen, die LQT1, LQT2 und LQT3 verursachen: KCNQ1, KCNH2 und SCN5A, welche jeweils gen-spezifische Auslöser wie körperliche Anstrengung (LQT1), emotionalen Stress (LQT2) und Schlaf (LQT3) aufweisen. Die genetische Untersuchung identifiziert eine Mutation bei 75% der LQTS-Fälle, wobei die drei Hauptgene für 90% der positiv genotypisierten Fälle verantwortlich sind [9]. LQT1-Patienten entwickeln meist bereits in der Kindheit Symptome und sind überwiegend männlich, während LQT2- und LQT3-Patienten Symptome etwas später, meist in der Pubertät, entwickeln und überwiegend weiblich sind [9].
Fallbericht
Wir präsentieren einen Fallbericht über eine schwangere Patientin mit einem zuvor diagnostiziertem LQT2 und erläutern die angewendeten Behandlungsstrategien für eine sichere Schwangerschaft, Entbindung und postpartale Phase.
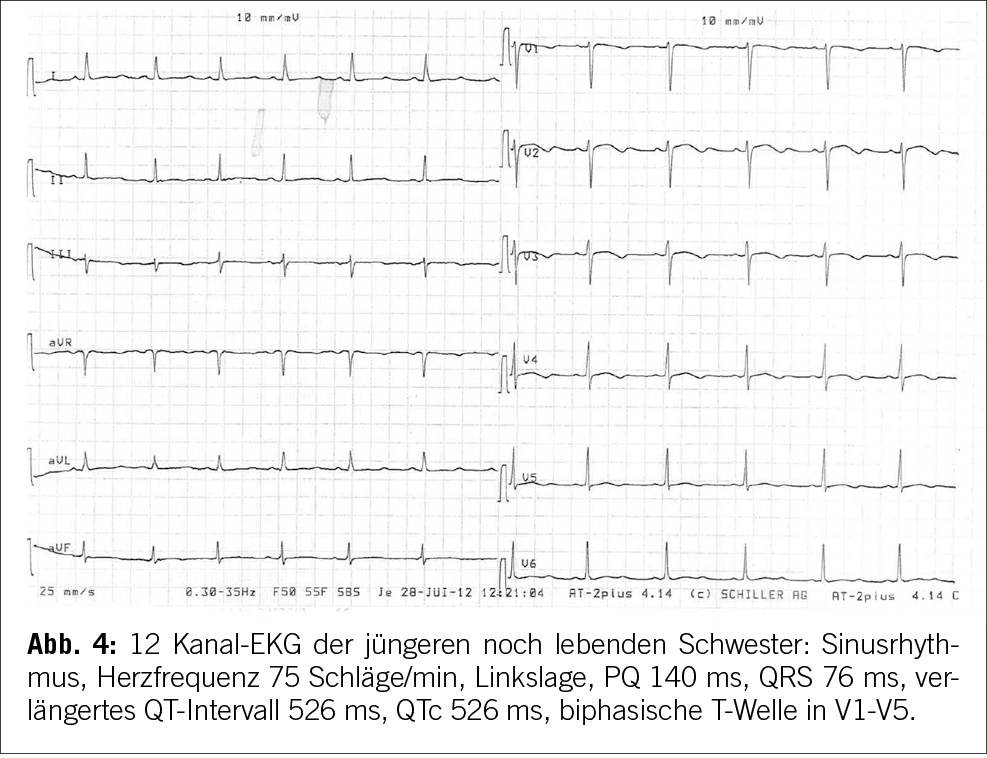
Unsere Patientin hatte zwei Schwestern, von denen eine im Jahr 2012 im Alter von 17 Jahren plötzlich in einem Skilager in den Anden im Schlaf verstarb. 3 Monate vor ihrem Tod erlitt die Schwester unserer Patientin eine Synkope. Die Synkope ereignete sich während des Tages beim Aufstehen vom Mittagstisch, die Schwester verlor plötzlich das Bewusstsein. Während dieses Vorfalls knirschte sie mit den Zähnen, atmete nicht und zeigte tonisch-klonische Bewegungen, die insbesondere die oberen Gliedmaßen betrafen. Offenbar hatte sie keinen tastbaren Puls, aber nach 2 externen Beatmungen erlangte sie das Bewusstsein zurück und erholte sich rasch. Sie wurde in einem örtlichen Krankenhaus in der Schweiz untersucht und ein EKG zeigte eine ausgeprägte Verlängerung des QT-Intervalls von über 500 ms und morphologische Abnormalitäten der T-Welle in mehreren Ableitungen. Ein Holter-Monitor zeigte ebenfalls eine Verlängerung des QT-Intervalls und morphologische Abnormalitäten der T-Welle. Es wurde jedoch keine Diagnose gestellt, wenige Monate danach verstarb die Schwester.
Es wurde eine Autopsie durchgeführt, die jedoch unauffällig ausfiel. Eine molekulare Autopsie ergab eine pathogene heterozygote Variante in KCNH2: Trp100X, die Long QT Typ 2 verursacht und den plötzlichen Herzstillstand erklärt. Eine kaskadenartige genetische Untersuchung ergab, dass die Patientin und ihre noch lebende, jüngere Schwester ebenfalls heterozygote Träger der Variante KCNH2: Trp100X sind. Beide wurden mit Nadolol behandelt. Auch die beiden Eltern wurden getestet. Die Mutter war genetisch negativ, während der Vater positiv für Mosaikismus von KCNH2: Trp100X war. Er zeigte jedoch keine phänotypischen Merkmale.
Unsere Patientin wurde zunächst mit 40 mg/Tag Nadolol behandelt, aber die Medikation wurde aufgrund von Müdigkeit abgesetzt. Nach dem Absetzen der Betablocker-Therapie hatte sie einen „epileptiformen“ Synkopen-Anfall, der durch das Klingeln eines Weckers ausgelöst wurde. Daher wurde Nadolol in einer reduzierten Dosis von 20 mg/Tag erneut verordnet. Seitdem war die Patientin beschwerdefrei. Nach der Synkope wurde ein Event-Rekorder implantiert, um potentielle weitere ventrikuläre Rhythmusstörungen aufzeichnen und die Therapie entsprechend anpassen zu können.
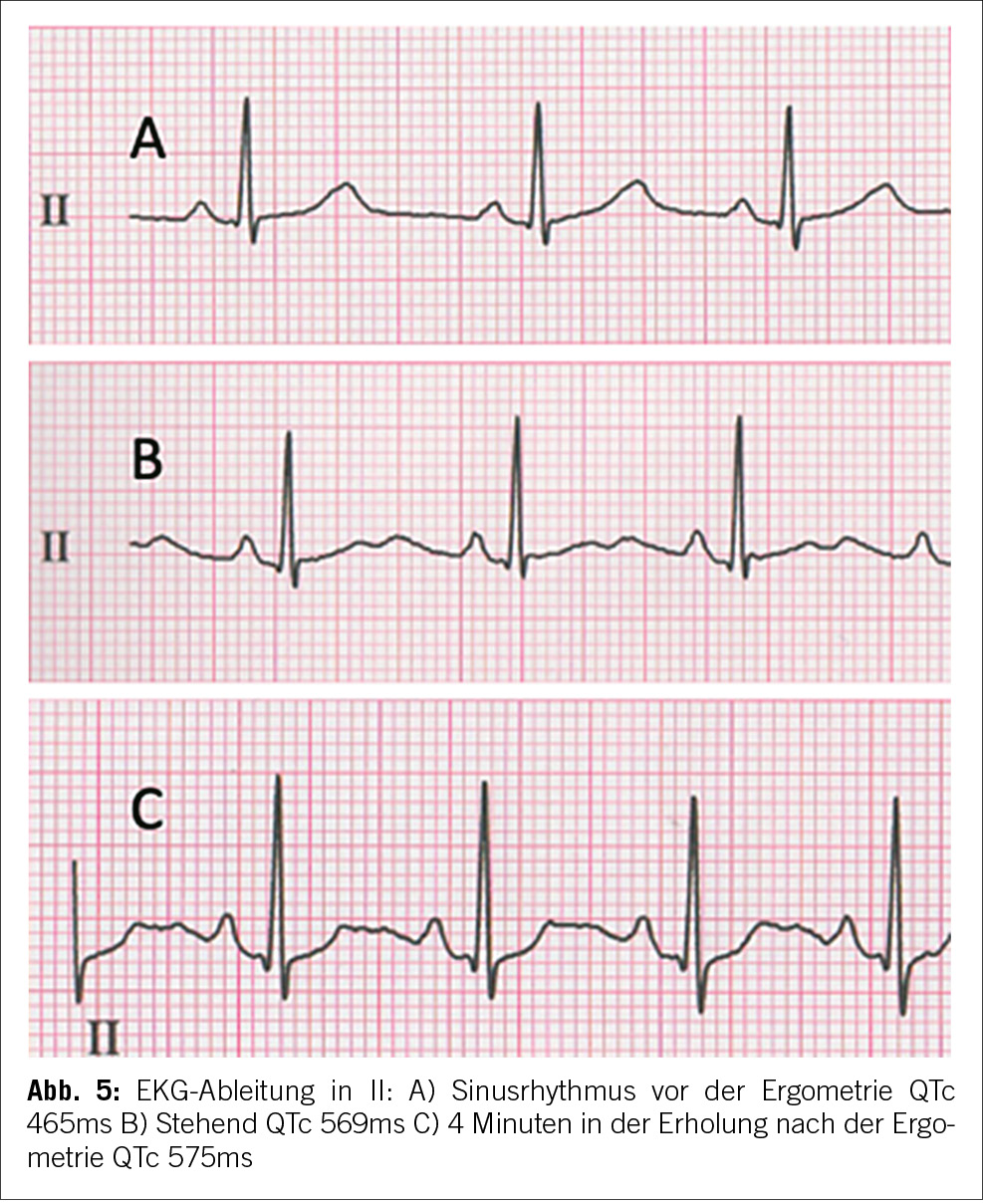
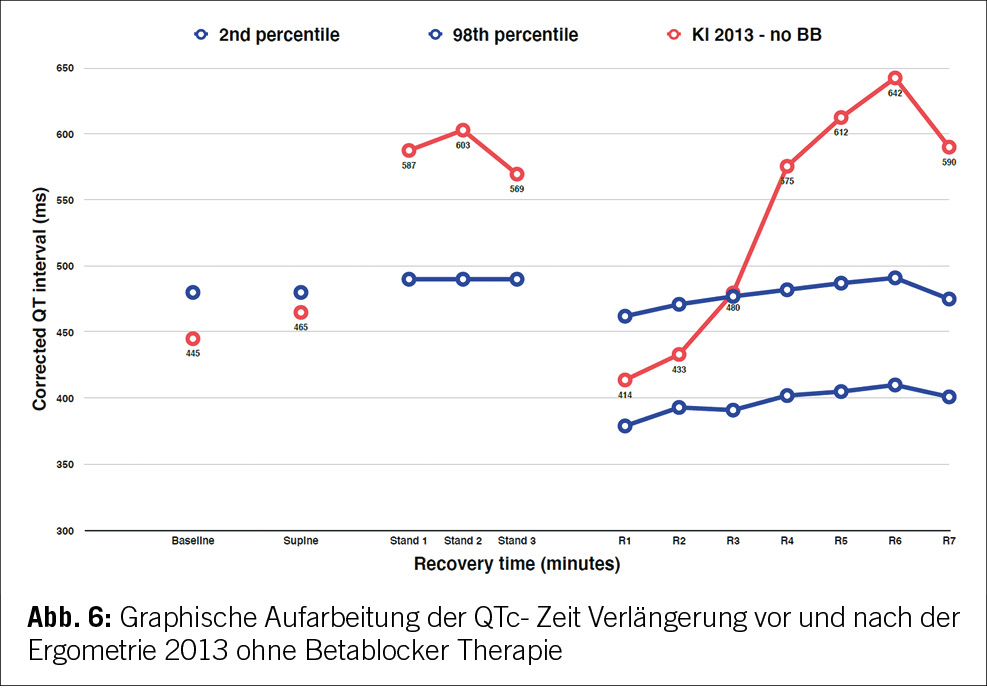
Die Patientin wies eine normale QT-Zeit im Ruhezustand auf. Signifikante QT-Zeit Verlängerungen und T-Wellen- Abnormalitäten traten beim Stehen und während der Erholung nach körperlicher Anstrengung auf. Dies deutete auf ein verborgenes Long-QT-Syndrom hin, das durch einfache Manöver manifest wurde. Die T-Wellen-Morphologie war mit einem Long-QT-Typ 2 vereinbar (Abbildung 5).
Verlauf der Schwangerschaft und postnatale Periode
Die Patientin zog im Alter von 29 Jahren in die Schweiz und äusserte einen Kinderwunsch. Es erfolgte ein Beratungsgespräch einige Monate vor der Schwangerschaft, welches die Risiken, die mit LQT2 während der Schwangerschaft verbunden sind, aufzeigte. Die Patientin wurde über die Wichtigkeit der täglichen Medikamenteneinnahme informiert. Nach dem Eintritt der Schwangerschaft wurde die Patientin alle 2-3 Monate mittels EKG und Elektrolytkontrollen in der kardiologischen Praxis kontrolliert. Der Betablocker wurde langsam auf die Zieldosis von Nadolol 1mg/kg (aufgeteilt auf eine Morgen- und Abenddosis) gesteigert, um in der für die Patientin vulnerabelsten Phase, während des Peripartum, die Zieldosis zu erreichen. Aufgrund der erwarteten Gewichtszunahme während der Schwangerschaft wurde die Dosis angepasst. Während der Verlaufskontrollen wurde der Event-Rekorder regelmässig abgefragt und zwischenzeitlich Daten remote übermittelt. Im 3. Trimester verspürte die Patientin Palpitationen, die durch den Event- Rekorder aufgezeichnet wurden. Hier zeigten sich in der Abfrage isolierte ventrikuläre Extrasystolen und eine ventrikuläre Salve über 4 Schläge. Ein gesundes Baby wurde vaginal entbunden, und die postnatale Phase unter Betablocker (Nadolol 1 mg/kg aufgeteilt auf eine Morgen- und Abenddosis) verlief ereignislos. Jedoch kam es 2 Monate nach der Entbindung unter optimaler Betablocker Dosierung zu einer nicht anhaltenden Torsade de Pointes Tachykardie. Die Betablocker Therapie wurde um 10 mg/Tag (von 80 mg auf 90 mg) erhöht und die Möglichkeit einer primärpräventiven ICD-Implantation mit der Patientin diskutiert. Die erhöhte Dosierung des Betablockers wurde erst nach der Aufteilung in 3 Tagesdosen toleriert. Ausserdem wurde die tägliche Einnahme von Magnesium erst nach Wechsel auf eine stärkere Verdünnung eingehalten. In den folgenden Monaten wurden keine Rhythmusstörungen verspürt bzw. vom Event-Rekorder aufgezeichnet. Daher wurde vorerst weiterhin von einer ICD-Implantation abgesehen.
Diskussion
Anhand dieses Falles lässt sich die Wichtigkeit eines Kaskadenscreenings nach diagnostiziertem Index bei Patienten darstellen. Nach der korrekten Diagnosestellung sowie genetischer Untersuchung und Beratung, ist die weitere kardiologische Begleitung in allen Lebenslagen notwendig.
Behandlungsstrategien
Die Grundlage der Behandlung von Patienten mit Long-QT-Syndrom (LQTS) stellt die Betablocker-Therapie dar. Entgegen der gängigen Meinung sind nicht alle Betablocker gleich wirksam. Die beiden effektivsten sind zweifellos Nadolol und Propranolol. Metoprolol und Atenolol sind weniger wirksam und sollten vermieden werden [5,6]. Daher werden ausschliesslich die nicht-selektiven Betablocker Nadolol und Propranolol als die wirksamsten Medikamente empfohlen [6].
Die neuen HRS Leitlinien von 2023 sprechen sich ausserdem klar für eine durchgehende Einnahme einer Betablockade bei schwangeren Patientinnen mit Long-QT-Syndrom aus. Diese sollte auch in der postpartalen Phase einschließlich des Stillens als Zeit mit erhöhtem Risiko für kardiale Ereignisse fortgesetzt werden (Evidenz Grad I, Empfehlungsgrad B) [12]. Propanolol wird dabei deutlich weniger in der Muttermilch ausgeschieden als Nadolol [13]. Jedoch zeigte eine Studie bei 68 Lebendgeburten von 31 Frauen mit LQTS der Cleveland Clinic in 2022, dass die Betablockade, insbesondere mit Nadolol, nicht mit einer höheren Inzidenz von intrauteriner Wachstumsretardierung assoziiert war. Darüber hinaus waren neonatale Bradykardien selten und Hypoglykämien wurde nicht beobachtet [14].
Nadolol hat eine längere Halbwertszeit als Propanolol, was anstatt einer dreimaligen eine zweimalige tägliche Einnahme ermöglicht, normalerweise in einer Dosierung von 1 bis 1,5 mg/kg pro Tag. Propranolol sollte in einer Dosierung von 2 bis 3 mg/kg pro Tag, verabreicht werden [5]. Metoprolol und Atenolol sind weniger wirksam und sollten vermieden werden. Die antiarrhythmische Wirkung der Betablocker bei LQTS beruht darauf, sogenannte frühe Nachdepolarisationen zu verhindern, indem sie den Einstrom von Kalzium-ionen reduzieren. Nadolol ist in der Schweiz nicht erhältlich und muss nach einer Kostengutsprache oder auf eigene Kosten aus dem Ausland importiert werden.
Betablocker Nebenwirkungen und Stillzeit
Bei Schwangerschaften, in denen die Mutter einen Beta-blocker einnimmt, ist es notwendig, ein Monitoring auf eine intrauterine Wachstumsretardierung des Fötus während der Schwangerschaften durchzuführen [12]. Bei Neugeborenen, die in utero mit Betablockern behandelt wurden, besteht potentiell ein Risiko für postnatale Symptome einer Betablockade wie Hypoglykämie und Bradykardien. Die Betablockade hemmt die Glykogenolyse, die durch die Aktivierung des sympathischen Nervensystems verursacht wird. Insbesondere anhaltende Hypoglykämien können bei Neugeborenen schwere Hirnverletzungen verursachen, daher ist es wichtig, auch das Neugeborene im Wochenbett und während der Stillzeit regelmässig zu überwachen [13]. Sollte dies unter Nadolol der Fall sein, müsste ggf. auf Propanolol bei der Mutter gewechselt werden, sollte das Stillen weiter gewünscht sein. Der Schutz der Mutter vor Rhythmusstörungen ist dann etwas geringer [13]. Der Kinderarzt sollte im Voraus informiert werden und Mutter und Kind sollten nach der Entbindung 5 Tage im Krankenhaus zur Überwachung verbleiben [1]. Wenn das Kind jedoch ebenso von einem Long-QT-Syndrom betroffen ist, hat es durch die Einnahme der Mutter von Nadolol ebenso eine Behandlung bis zur Geburt bzw. in der Stillzeit.
Weitere Behandlungsstrategien
Eine weitere, eskalierende Therapieform besteht in der linkskardialen sympathischen Denervation (Entfernung der ersten 3 bis 4 Ganglia thoracica idealerweise durchgeführt via extrapleuralem Zugang, der einen Thorakotomie-Eingriff unnötig macht) des linken herznahen sympathischen Nervensystems (LCSD), was eine Alternative bei Betablocker-resistenten Patienten darstellt. Tatsächlich hat LCSD keinen negative Einfluss auf die kardiovaskuläre Leistung [3]. Der Expertenkonsens der HRS/EHRA/APHRS empfiehlt, LCSD bei Hochrisiko-Patienten mit der Diagnose LQTS durchzuführen, wenn die ICD-Therapie kontraindiziert ist, abgelehnt wird oder trotz maximal tolerierter kombinierter Therapie mit Betablocker nicht wirksam bei der Verhinderung von Synkopen/Arrhythmien ist. LCSD ist besonders wirksam bei LQTS1-Patienten [7].
Eine vaginale Geburt wird als eine sichere Entbindungsstrategie bei Patientinnen mit LQTS angesehen, wobei randomisierte Studien fehlen und die Empfehlungen auf Fallberichten und Expertenmeinungen fussen [2].
Die Schwangerschaft und die ersten 9 Monate nach der Geburt bei Patientinnen mit bekanntem LQTS bergen ein deutlich erhöhtes Risiko für lebensbedrohliche Arrhythmien [3]. Es wird angenommen, dass die Veränderungen in der adrenergen Aktivität in der peripartalen Phase zu einer Zunahme kardialer Ereignisse führen kann. Erklärend könnte die Zunahme der sympathischen Aktivität sein, die mit dem Stress und vor allem veränderten Schlafmuster bei der Betreuung eines Neugeborenen einhergeht. Diese könnte die ventrikulären Arrhythmien bei Patienten mit LQT1- und LQT2-Genotypen triggern [3]. Die Östrogen- und Progesteronspiegel sind während der Schwangerschaft hoch und fallen deutlich unter die normale Werte, wenn die Mutter ihr Kind stillt. Diese Veränderung der Hormonspiegel könnte die adrenergen Reaktionen der mutierten Ionenkanäle bei LQTS beeinflussen [10].
LQTS-Patientinnen bedürfen rund um die Schwangerschaft einer engmaschigen kardiologischen Verlaufskontrolle, um potentiell lebensbedrohliche Rhythmusstörungen durch eine gute Betablocker Einstellung weitestgehend zu verhindern. Eine Schwangerschaft ist jedoch möglich und sollte bei entsprechendem Patientinnen-Wunsch, wenn immer medizinisch vertretbar, befürwortet werden. In diesem Fallbericht wird die Herausforderung der Behandlung der Patientin – auch bei Nebenwirkungen des Betablockers –
beleuchtet. Die Therapie regelmässig an das Gewicht anzupassen ist gerade in der Schwangerschaft und postnatalen Phase essentiell.
Die Entbindung sollte in einem Spital erfolgen und es müssen unmittelbare Wiederbelebungsmassnahmen verfügbar sein, einschließlich eines externen Defibrillators [6,11]. Es wird empfohlen, frühzeitig einen Anästhesisten hinzuzuziehen, um eine sichere Analgesie während der Wehen und der Geburt zu planen. Die Entscheidung über eine Epiduralanästhesie und eine assistierte Entbindung sollte auf der Grundlage einer geburtshilflichen Indikation getroffen werden; diese Eingriffe sind aufgrund eines vorliegenden LQTS nur selten indiziert. Während der Entbindung ist es wichtig, Umstände zu vermeiden, die das Risiko für ventrikuläre Arrhythmien erhöhen könnten wie z. B.: Elektrolytstörungen, starke Blutungen oder QT-Zeit verlängernde Medikamente (siehe diesbezüglich crediblemeds.org). Bei Patientinnen mit LQTS wird empfohlen, in einer ruhigen Umgebung zu entbinden. Das Neugeborene hat, aufgrund der autosomal dominanten Vererbung, ein Risiko von 50%, ebenfalls von einem LQTS betroffen zu sein. Die frühzeitige Diagnose ist bei Kindern wichtig, denn sie hat therapeutische Konsequenzen. Eine genetische Testung des Neugeborenen kann via Blut aus der Nabelschnur durchgeführt werden. Diesbezüglich sollte am besten vor der Schwangerschaft eine genetische Beratung stattfinden.
Schlussfolgerung
Ein Long-QT-Syndrom stellt keine Kontraindikation für eine Schwangerschaft dar, das Risiko von Herzrhythmusstörungen wird durch eine Therapie mit Betablockern drastisch reduziert. Bei Patientinnen mit LQTS ist eine vaginale Entbindung möglich, eine Hausgeburt wird nicht empfohlen [1]. Eine regelmässige kardiologische Mitbetreuung ist unabdingbar, bezüglich des Kontrollintervalls ist eine kardiologische Konsultation alle 4-6 Wochen sinnvoll, eventuell ergänzt durch das Monitoring eines Event-Rekorders.
Abkürzungen
LQTS = Long-QT-Syndrom
LQT1 = Long–QT-Syndrom 1 (Genotyp 1 mit einer Mutation im KCNQ1-Gen)
LQT2 = Long-QT-Syndrom 2 (Genotyp 2 mit einer Mutation im KCNH2-Gen)
LQT3 = Long-QT-Syndrom 3 (Genotyp 3 mit einer Mutation im SCN5A -Gen)
EKG = Elektrokardiogramm
SCD = sudden cardiac death, plötzlicher Herztod
QT = Zeitintervall vom Anfang des QRS-Komplexes bis zum Ende der
T-Welle
QTc = frequenzkorrigierte QT- Zeit (in diesem Artikel nach Bazett- Formel)
ICD = Implantierbarer Kardioverter-Defibrillator
LCSD = linkskardiale sympathische Denervation
HRS = Heart Rhythm Society: Die Heart Rhythm Society
EHRA = European Heart Rhythm Association
APHRS = Asia Pacific Heart Rhythm Society
Adlerstrasse 1
8600 Dübendorf
Es bestehen keine Interessenskonflikte.
Historie:
Manuskript eingereicht: 03.11.2023
Nach Revision angenommen: 06.12.2023
1. Asatryan B, Rieder M, Castiglione A, Odening KE. Arrhythmic risk during pregnancy and postpartum in patients with long QT syndrome. Herzschrittmacherther Elektrophysiol. 2021 Jun;32(2):180-185. doi: 10.1007/s00399-021-00757-4. Epub 2021 Mar 29. PMID: 33782754; PMCID: PMC8166676.
2. Seth R, et al. Long QT syndrome and pregnancy. J Am Coll Cardiol. 2007; 49(10):1092-1098
3. Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Executive summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society [published correction appears in Heart Rhythm. 2018 Sep 26;:]. Heart Rhythm. 2018;15(10):e190-e252. doi:10.1016/j.hrthm.2017.10.035.
4. Kaufman ES, Eckhardt LL, Ackerman MJ, et al. Management of Congenital Long-QT Syndrome: Commentary From the Experts. Circ Arrhythm Electrophysiol. 2021;14(7):e009726. doi:10.1161/CIRCEP.120.009726.
5. Wilde AAM, Semsarian C, Márquez MF, et al. EHRA/HRS/APHRS/LAHRS Expert Consensus Statement on genetic testing for cardiac diseases. EP Europace. 2022;24(8):1307-1367. DOI: 10.1093/europace/euac030.
6. Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management [published correction appears in Circ Arrhythm Electrophysiol. 2012 Dec;5(6):e119-20]. Circ Arrhythm Electrophysiol. 2012;5(4):868-877. doi:10.1161/CIRCEP.111.962019.
7. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2013;10(12):1932-1963. doi: 10.1016/j.hrthm.2013.05.014.
8. Marcinkeviciene A, et al. Long QT Syndrome Management during and after Pregnancy. Medicina (Kaunas, Lithuania). 2022 Nov 21;58(11):1694. doi:10.3390/medicina58111694.
9. Wilde AAM, Amin AS, Postema PG. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart. 2022;108(5):332-338. doi:10.1136/heartjnl-2020-318259.
10. Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, Blomström-Lundqvist C, Cífková R, De Bonis M, Iung B, Johnson MR, Kintscher U, Kranke P, et al. 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy. Eur Heart J. 2018; 39:3165–3241.
11. Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA, Charron P, Corrado D, Dagres N, de Chillou C, Eckardt L, Friede T, Haugaa KH, Hocini M, Lambiase PD, Marijon E, Merino JL, Peichl P, Priori SG, Reichlin T, Schulz-Menger J, Sticherling C, Tzeis S, Verstrael A, Volterrani M; ESC Scientific Document Group. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022 Oct 21;43(40):3997-4126. doi: 10.1093/eurheartj/ehac262. PMID: 36017572.
12. Oglar JA, Kapa S, Saarel EV, Dubin AM, Gorenek B, Hameed AB, Lara de Melo S, Leal MA, Mondésert B, Pacheco LD, Robinson MR, Sarkozy A, Silversides CK, Spears D, Srinivas SK, Strasburger JF, Tedrow UB, Wright JM, Zelop CM, Zentner D. 2023 HRS expert consensus statement on the management of arrhythmias during pregnancy. Heart Rhythm. 2023;20(10):e175–e264.
13. Martinez A, Lakkimsetti M, Maharjan S, Aslam MA, Basnyat A, Kafley S, Reddy SS, Ahmed SS, Razzaq W, Adusumilli S, Khawaja UA. Beta-Blockers and Their Current Role in Maternal and Neonatal Health: A Narrative Review of the Literature. Cureus. 2023;15(8):e44043.
14. Hammond BH, El Assaad I, Herber JM, Saarel EV, Cantillon D, Aziz PF. Contemporary maternal and fetal outcomes in the treatment of LQTS during pregnancy: Is nadolol bad for the fetus? Heart Rhythm. 2022;19(9):1516–1521.