
Die kardiale Amyloidose ist durch Ablagerungen von Amyloid bedingt, die häufigsten Formen sind monoklonale Leichtketten (AL) oder Transthyretin (TTR). Beide Formen können das Herz befallen und sind mit einer ungünstigen Prognose vergesellschaftet. Die kardiale TTR Amyloidose wird aufgrund der modernen diagnostischen Möglichkeiten immer häufiger diagnostiziert und ist viel häufiger als ursprünglich angenommen. Dies ist wichtig, da seit kurzem eine Therapie verfügbar ist, welche die Prognose verbessert.
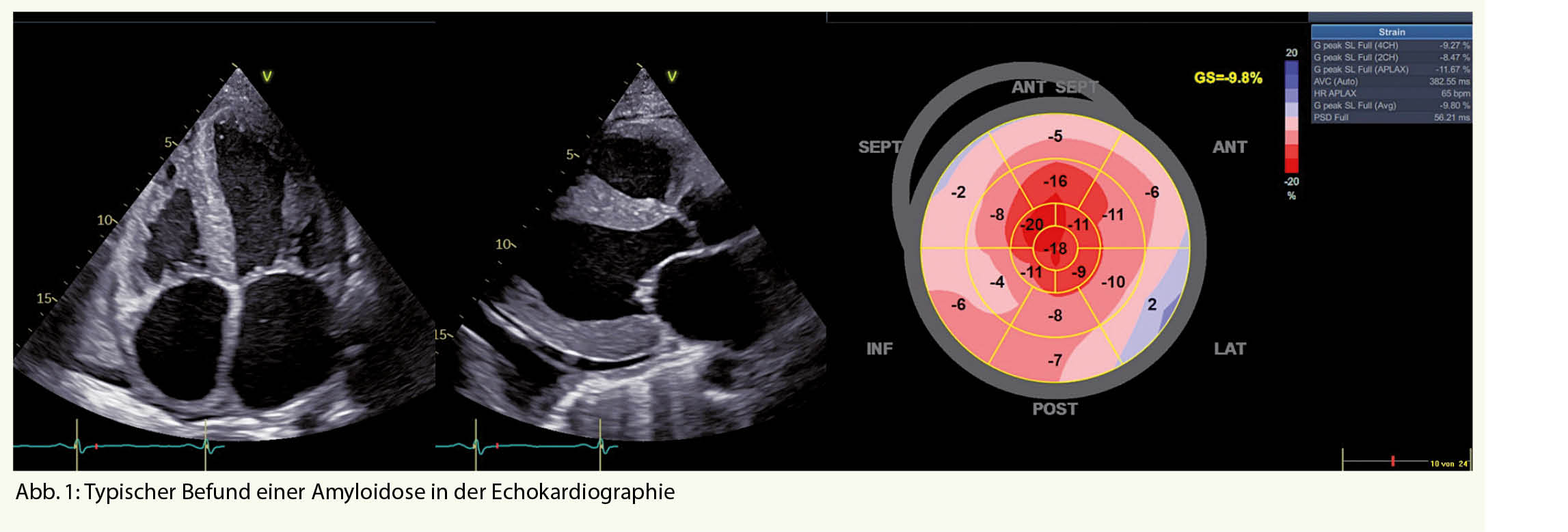
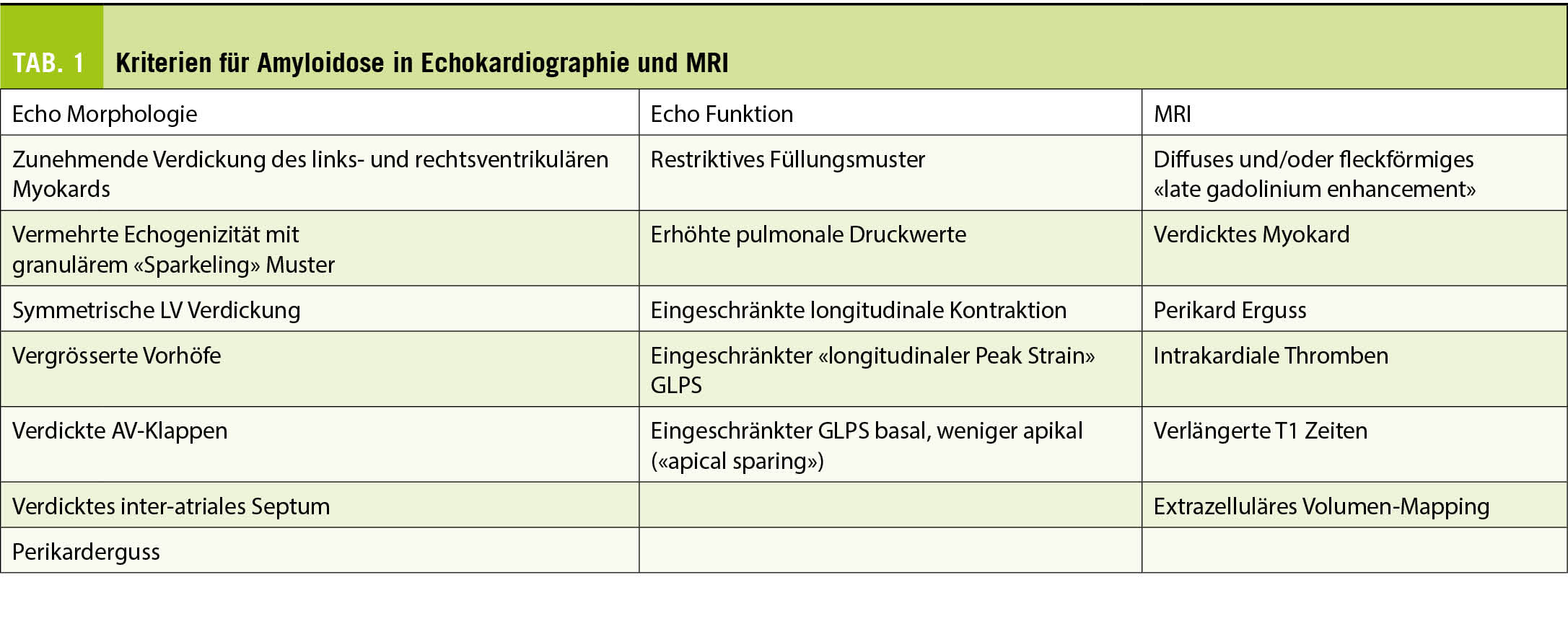
An eine kardiale Amyloidose sollte immer dann gedacht werden, wenn sich ein Patient mit Symptomen einer Herzinsuffizienz präsentiert und einen Echokardiographie- oder MRI-Befund aufweist der typisch für eine Amyloidose ist (Abb. 1 und Tab. 1) (1).
Bei der AL Amyloidose liegt die Ursache bei einer hämatologischen Erkrankung (monoklonale Gammopathie, meist Plasmazelldyskrasie, seltener lymphoplasmozytische Erkrankung) – hierbei bilden monoklonale Plasmazellen Leichtketten, welche sich im Herz und anderen Organen ablagern. Bei der TTR Amyloidose unterscheidet man die «wild-type (wt)» von der «variant» (genetische) Form. Bei der familiären Form liegt ein Gendefekt im TTR-Gen zugrunde, es kommt zu einer Fehlfaltung von TTR, das sich als Amyloid im Herzen, aber auch im Nervensystem ablagern kann (2). Der Pathomechanismus bei der wt-Form ist nicht abschliessend geklärt, wobei sich auch dort TTR als Amyloid in den Organen ablagert. Neben der kardialen Manifestation und insbesondere bei der wt-Form kann es typischerweise ein paar Jahre vor dem Herzbefall zum Karpaltunnel Syndrom, einer Bizeps-Sehnen-Ruptur oder einer Spinalkanalstenose kommen (3).
Die AL-Amyloidose ist eine seltene Erkrankung und hat immer noch eine schlechte Prognose, insbesondere wenn das Herz betroffen ist – neue Therapien haben aber hier deutliche Fortschritte erzielt. Auch die vATTR Amyloidose ist sehr selten, in gewissen Gebieten aber endemisch (z.B. Teilen von Portugal, Schweden oder Japan). Die wtATTR Amyloidose wurde bisher ebenfalls als seltene Erkrankung angesehen, in den letzten Jahren hat sich jedoch dank verbesserter Diagnostik gezeigt, dass diese Form viel häufiger ist und meist eine bessere Prognose aufweist als bisher angenommen. Für diese Form ist seit kurzem erstmals eine spezifische Therapie zugelassen.
Diagnostik
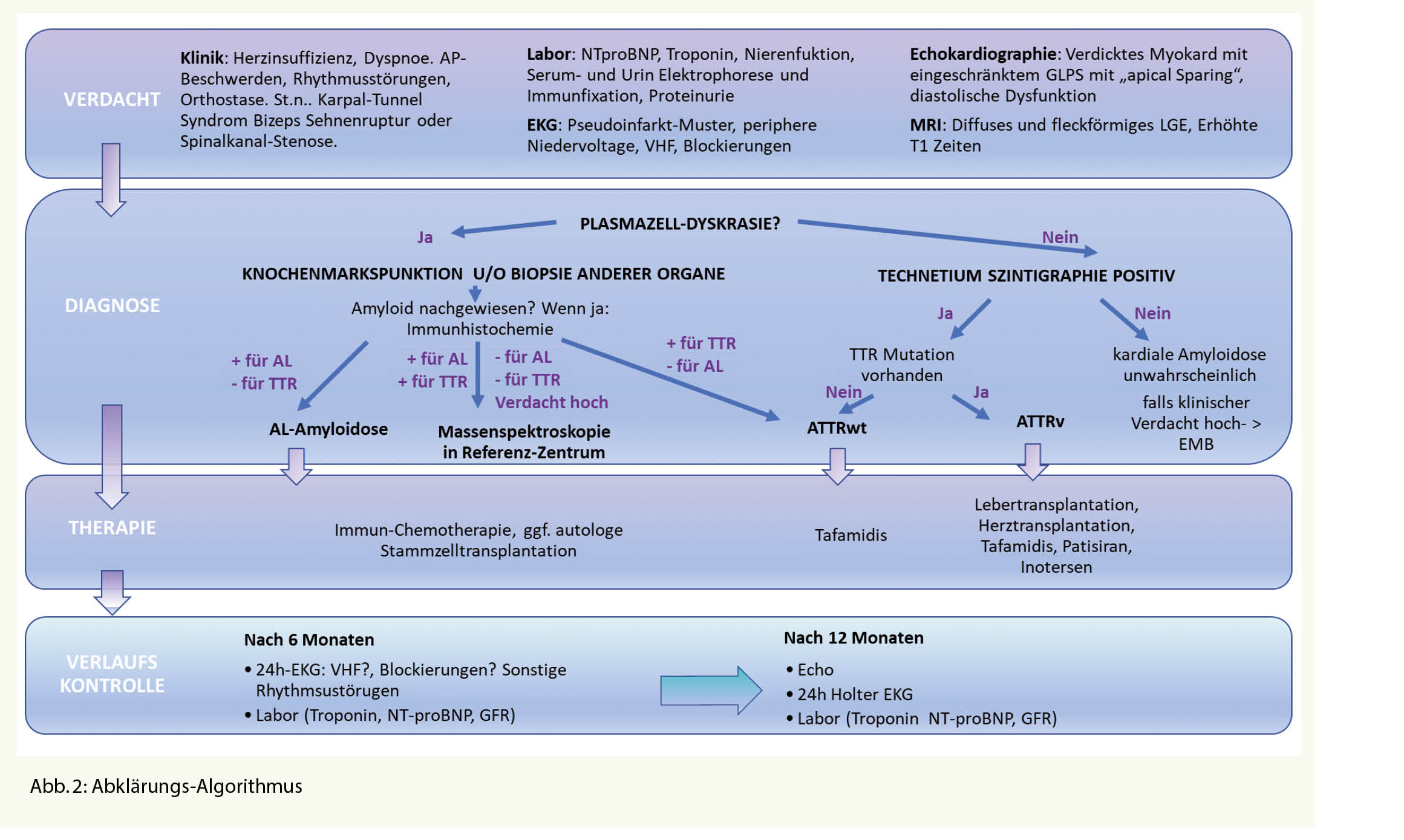
Bei Verdacht auf eine kardiale Amyloidose (Herzinsuffizienz-Beschwerden, typisches Echo oder MRI) sollten zuerst ausführliche Laboruntersuchungen inkl. kardiale Biomarker NT-pro BNP und Troponin (zur Abschätzung der Prognose) und eine Protein-Elektrophorese mit Immunfixation im Serum und Urin sowie Bestimmung der freien Leichtketten im Serum veranlasst werden. Letzteres ist entscheidend für die Unterscheidung zwischen AL und ATTR Amyloidose. Ein EKG sollte geschrieben werden, um die Frage nach Vorhofflimmern und Blockierungen zu beantworten. Die typischerweise beschriebene «low-voltage» findet sich eher bei der AL-Amyloidose, bei der ATTR ist dies jedoch nicht so häufig (ca. 22%), typischer ist dort ein Pseudoinfarktmuster (63%).
Bei unauffälliger Immunfixation ist eine AL-Amyloidose praktisch ausgeschlossen (Abb. 2). In diesem Fall sollte eine Technetium Szintigraphie durchgeführt werden. Fällt diese positiv aus, gilt die Diagnose einer ATTR-Amyloidose als gesichert (4), jedoch kann nicht zwischen wtATTR und vATTR unterschieden werden, weswegen eine genetische Testung sinnvoll ist. Diese kann nach Einholen einer Kostengutsprache durch die Krankenkasse durchgeführt werden kann. An eine hereditäre Form sollte insbesondere dann gedacht werden, wenn neurologische Symptome wie Polyneuropathie vorhanden sind. Bei negativer Szintigraphie und persistierend hohem Verdacht auf eine kardiale Amyloidose, sollte eine Endmyokardbiopsie durchgeführt werden (5).
Bei pathologischer Immunfixation und Verdacht auf eine kardiale Amyloidose ist die Situation etwas komplizierter, da die Diagnose mittels Biopsie und Nachweis von Amyloid gestellt werden muss. Normalerweise erfolgt bei pathologischer Immunfixation eine hämatologische Abklärung, gelegentlich gelingt der Nachweis von Amyloid schon in der Knochenmarksbiopsie (ca. 50%), ansonsten muss Amyloid an anderen Stellen (typischerweise Bauchfett, Speicheldrüsen, Endomyokard) gesucht und immunhistochemisch bestätigt werden, was gelegentlich herausfordernd sein kann (Abb. 2). Ein wichtiges Problem stellt die hohe Koinzidenz eines MGUS mit der wtATTR Amyloidose im fortgeschrittenen Alter dar.
Amyloidose mit Herzbefall
Während die AL-Amyloidose meist verschiedene Organe befällt (insb. Herz, Niere, Nervensystem und Magen-Darmtrakt), ist bei der wtATTR Amyloidose meist das Herz im Zentrum (bei der vATTR zusätzlich das Nervensystem). Was muss man besonders beachten bei einem Herzbefall? Im Zentrum steht die Herzinsuffizienz, mit den klinischen Zeichen einer restriktiven Kardiomyopathie (links- und rechtsseitige Volumenretention). Sehr wichtig ist die Neigung zu Vorhofflimmern und Hirninfarkten – ca. 90% der Patienten haben oder entwickeln im Verlauf ein VHF. Weitere Probleme sind Reizleitungsstörungen, welche insbesondere bei der ATTR-Amyloidose auftreten, sowie Synkopen und plötzlicher Herztod. Synkopen bei Anstrengung sind meist dadurch bedingt, dass das Herzminutenvolumen fixiert ist – die Hypotonie beziehungsweise die Orthostase aufgrund einer autonomen Dysfunktion. Auch Thoraxschmerzen aufgrund einer Beteiligung der Mikrozirkulation sind nicht selten.
Behandlung der kardialen Amyloidose
Für den Patienten steht initial die symptomatische Therapie im Vordergrund. Am wichtigsten ist die Behandlung mit Diuretika, um den Patienten von den Symptomen der Volumenretention zu entlasten. Hierbei gibt es aber zu bedenken, dass die Druck-Volumen-Kurven sehr steil sind und zu viel Diuretika auch einen übermässigen Blutdruck-Abfall bewirken können. Trotzdem müssen die Diuretika genügend hoch dosiert werden um den Patienten wirklich zu entlasten. Im Verlaufe der Erkrankung sind meist immer höhere Dosen notwendig. Die Erkrankung hat einen HFpEF Charakter und die Auswurffraktion ist meist «normal.» Das bedeutet, dass trotz Herzinsuffizienz-Symptomen keine Beta-Blocker, ACE-Hemmer (ACHI), Angiotensin-Rezeptor-Blocker (ARB) oder gar Sacubitril/Valsartan verabreicht werden sollten. Beta-Blocker werden meist schlecht vertragen da das Herzminutenvolumen kaum mehr gesteigert werden kann (das Schlagvolumen ist fixiert und der kardiale Output kann nur noch mit der Herzfrequenz gesteigert werden). Vor allem bei der AL-Amyloidose, aber auch bei der fortgeschrittenen TTR-Amyloidose werden ACEI und ARB aufgrund ihrer peripher vasodilatierenden Wirkung sehr schlecht vertragen und können Orthostase und Synkopen verursachen.
Von grösster Wichtigkeit ist das Erkennen von Vorhofflimmern und eine rechtzeitige orale Antikoagulation, um den Patienten vor einem Schlaganfall zu bewahren (häufige Erstmanifestationen einer Amyloidose). Wir empfehlen deshalb ein Holter-EKG alle 6 Monate durchzuführen. Bei einem dokumentieren VHF sollte unabhängig vom CHA2DS2-VASc Score eine orale Antikoagulation (OAK) begonnen werden. Einige Zentren beginnen damit schon, wenn der Patient noch im Sinus-Rhythmus ist, jedoch in der Echokardiographie ein restriktives Füllungsmuster aufweist. Die Rationale dahinter ist, dass in solchen Fällen die intraatrialen Druckwerte so hoch sind, dass kaum mehr Fluss erzeugt wird und der Patient zu Thromben neigt. Daten für Vorhofflimmern-Ablationen in der Amyloidose sind kaum vorhanden, wir sind hier zurückhaltend.
Die Indikationen für einen Herzschrittmacher unterscheiden sich nicht wesentlich von Patienten ohne Amyloidose, eine relevante Blockierung ist jedoch häufiger. Eine der grössten Schwierigkeiten ist die Prävention des plötzlichen Herztods. ICD-Implantation müssen im Team und mit dem Patienten gut besprochen werden und der Nutzen und die Risiken gegeneinander abgewogen werden. Es gilt zu bedenken, dass Fälle von elektromechanischer Entkoppelung und nicht Kammerflimmern die häufigsten Ursachen für einen rhythmogenen Tod bei Amyloidose darstellen – ein ICD hilft hier nicht. Auch sind inadäquate (und adäquate) Schocks häufiger als bei Nicht-Amyloidose und die Wirksamkeit eines Schocks kann aufgrund der Amyloid-Ablagerungen auch eingeschränkt sein.
Eine ATTR-Amyloidose wird immer häufiger bei Patienten mit Aortenstenose, welche für eine TAVI abgeklärt werden, festgestellt. Hier stellt sich die Frage ob man besser die Amyloidose oder die Aortenstenose behandelt. Es konnte gezeigt werden, dass eine TAVI bei diesen Patienten sicher ist und den Patienten nicht aufgrund der kardialen Amyloidose vorenthalten werden sollte (6).
In seltenen Fällen kommt auch die Herztransplantation in Frage. Hier ist es jedoch so, dass insbesondere bei der AL-Amyloidose die Patienten häufig bereits einen Multiorganbefall aufweisen, was eine relative Kontraindikation für eine Transplantation darstellt. Das Ausmass der systemischen Beteiligung ist hierbei entscheidend. Selten kann man bei einem isolierten Herzbefall eine Transplantation durchführen um anschliessend eine intensive Chemotherapie mit autologer Stammzelltransplantation zu ermöglichen. Bei der ATTR Amyloidose mit rein kardialem Befall ist eine Transplantation in einigen Fällen gut möglich, meist haben die Patienten das TPL-Alter jedoch bereits überschritten.
Behandlung der zugrundeliegenden Erkrankung
Bei der AL-Amyloidose richtet sich die Therapie gegen die zugrundeliegende Erkrankung. Hier hat es in den letzten Jahren grosse Fortschritte gegeben, welche hier nicht im Detail besprochen werden. Wichtig ist die Zusammenarbeit mit einem für die Amyloidose spezialisierten Zentrum, um den Patienten in einem interdisziplinären Umfeld optimal zu betreuen und auch die neusten Therapien im Rahmen von Studien zugänglich zu machen.
Auch bei der ATTR-Amyloidose gab es in den letzten Jahren mehrere entscheidende Durchbrüche in der Behandlung. Mit Patisiran (RNA-Interferenz) und Inotersen (RNA antisense Oligonukleotid) kann die Bildung von Transthyretin in der Leber fast ganz blockiert werden (7). Zwei Studien bei Patienten mit einer familiären Amyloid-Polyneuropathie (familiär bedingte ATTR-Amyloidose welche das periphere Nervensystem befällt) zeigen eindrücklich, wie die Polyneuropathie in vielen Fällen aufgehalten oder gar verbessert wurden – auch bezüglich kardialer Symptome gibt es erste Hinweise für einen Nutzen. Entsprechende Studien bei der kardialen Amyloidose (vATTR und wtATTR) werden aktuell durchgeführt.
Ein weiterer Meilenstein ist die Zulassung von Tafamidis, einem TTR Stabilisator, für die Behandlung der kardialen ATTR-Amyloidose. Die ATTR-ACT Studie (mittleres Alter ca. 74 Jahre, 90% Männer, 75% wtATTR) zeigte, dass die Mortalität und die kardiovaskulär bedingten Hospitalisierungen über die Studiendauer von 30 Monaten signifikant gesenkt werden konnte (8). Bis dieser Effekt eintrat, dauerte es jedoch einige Monate. Interessant ist auch, dass die Lebensqualität und die Gehstrecke unter Tafamidis bei einem günstigen Nebenwirkungsprofil (kaum Therapieabbrüche in der Studie) weniger schnell abnehmen. Die Medikation ist zwar jetzt in der Schweiz zugelassen, jedoch nicht auf der Spezialitätenliste zu finden. Ein Kostengutsprache-Gesuch muss deshalb zwingend gestellt werden, die Verfügbarkeit ist zur Zeit limitiert.
Copyright bei Aerzteverlag medinfo AG
LA Klinik für Kardiologie
Amyloidose-Netzwerk Zürich
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
LA Klinik für Kardiologie
Amyloidose-Netzwerk Zürich
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
LA Klinik für Kardiologie
Amyloidose-Netzwerk Zürich
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
andreas.flammer@usz.ch
NL: In Verbindung mit dem Manuskript: Honorare von Pfizer und Kongressbeiträge von Alnylam
RS: In Verbindung mit dem Manuskript: Honorare von Alnylam und Pfizer und einen ‘unrestricted research grant’ von Pfizer
Nicht in Verbindung mit dem Manuskript: Honorare oder Kongressbeiträge von Janssen, Mundipharma und Takeda
AFL: In Verbindung mit dem Manuskript: Honorare von Alnylam und Pfizer
Nicht in Verbindung mit dem Manuskript: Honorare oder Kongressbeiträge von AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Fresenius, Imedos Systems, Medtronic, Mepha, Mundipharma, Novartis, Roche, Schwabe Pharma, Vifor und Zoll
1. Brouwers S, et al.: Cardiac Amyloidosis
Cardiovasc Med. 2018;21(11):282-289
2. Rauch PJ et al.: Systemische Amyloidosen
Schweiz Med Forum 2014 14;943-048
3. Maurer MS et al: Expert Consensus Recommendation for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis
Circulation: Heart Failure 2019; 12
4. Gillmore JD et al.: Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis
Circulation 2016: j133(24):2404-12
5. Laptseva N et al.: Cardiac amyloidosis: still challenging
Eur Heart J 2017, 38(22):122
6 Scully PR et al: Prevalence and outcome of dual aortic stenosis and cardiac amyloid pathology in patients referred for transcatheter aortic valve implantation
EHJ, 2020; 41; 759-2767
7. Maurer MS et al.: Tafamidis Tratment for Patients with Transthyretin Amyloid Cardiomyopathy, N Engl J Med 2018; 379(11):1007-1016
8. Adams D et al.: Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis
N Engl J Med 2018; 379(1):11-21



der informierte @rzt
- Vol. 11
- Ausgabe 1
- Januar 2021