
Vaskulitiden sind in Ätiologie und Manifestation sehr mannigfaltig. Als Arteriitiden, also Grossgefässvaskulitiden, gelten die Riesenzellarteriitis und die Takayasu-Arteriitis, auf die der Artikel fokussiert. Die Polymyalgia rheumatica hat einen engen Zusammenhang mit der Riesenzellarteriitis. Arteriitiden können klinisch vermutet werden, eine bildgebende oder bioptische Sicherung ist obligat. In der Frühphase dominieren klinisch Entzündungssymptome, später können Durchblutungsstörungen auftreten. Verspätete Therapie kann fatale Folgen haben. Eckpfeiler der Behandlung bleiben Steroide. Alternative immunsupprimierende Medikamente wie Metho-trexat oder Tocilizumab reduzieren den Steroidverbrauch.
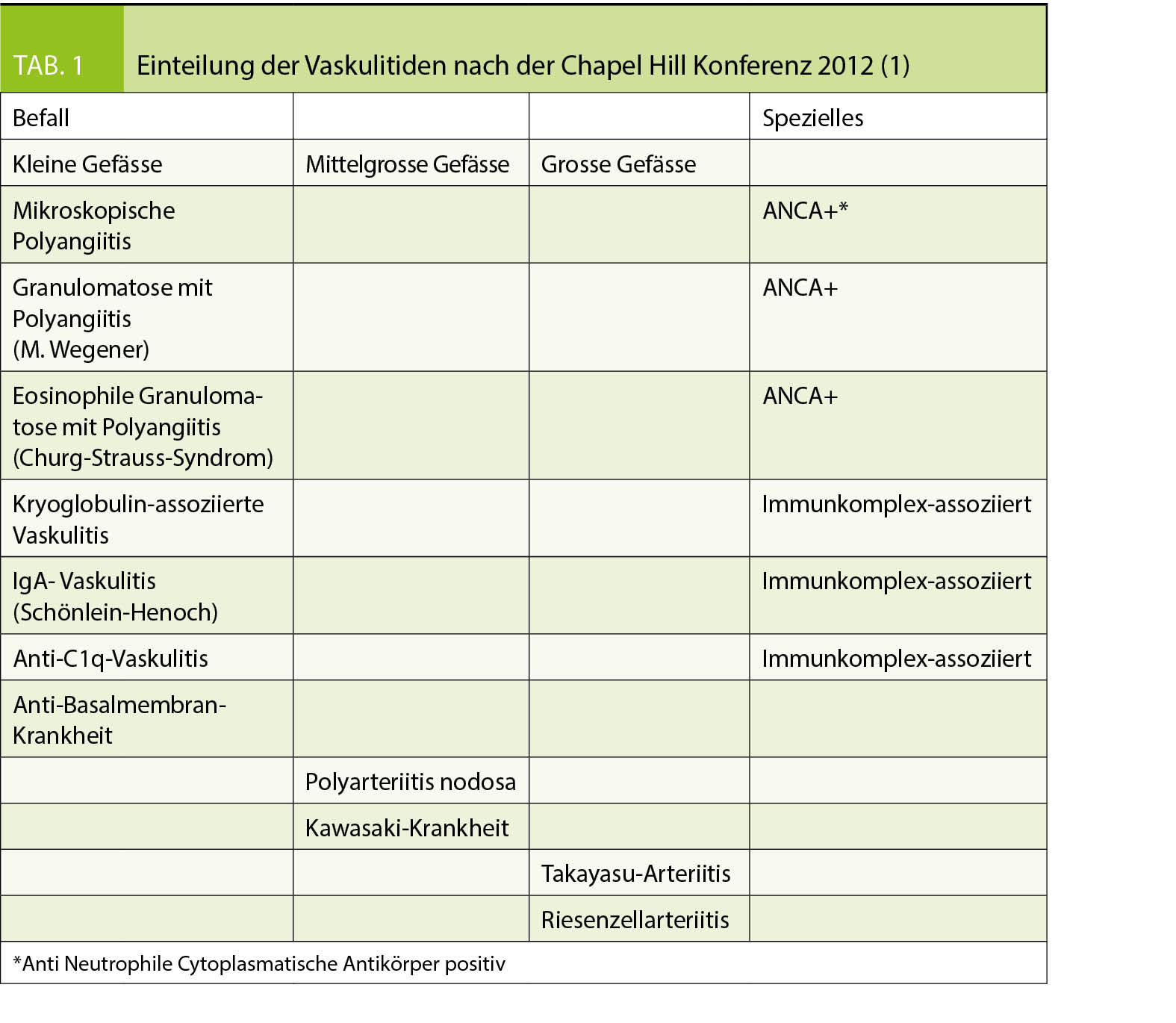
Arteriitiden sind Vaskulitiden der grossen Arterien. Die Chapel Hill Konferenz 2012 definierte eine auch heute noch gängige Einteilung der komplexen Krankheitsbilder (1). Tab. 1 zeigt diese Einteilung, die sich an der Grösse des befallenen Gefässes orientiert. Bioptisch finden sich bei Arteriitis mit Leukozyten, speziell auch Riesenzellen, infiltrierte Gefässwände. Initial aktiviert ein autoimmuner Mechanismus T-Lymphozyten. Die nachfolgende Entzündungskaskade führt unbehandelt zur Stenosierung oder zum Verschluss des betroffenen Gefässes oder zur Zerstörung der Wand mit Aneurysmabildung bis zur Ruptur.
Der vorliegende Artikel fokussiert auf die Riesenzellarteriitis (RZA) und die Takayasu-Arteriitis (TA). Die faszinierenden neuen Aspekte in der Abgrenzung von RZA und TA als auch der Stellung der Polymyalgia rheumatica (PMR) sollen kurz beleuchtet werden.
Zwei Krankheiten oder gar drei oder nur eine?
Die RZA, früher oft auch Arteriitis temporalis Horton genannt, die TA und die PMR wurden früher als 3 eigenständige Krankheitsbilder interpretiert. Diese Sicht ist in der Zwischenzeit in Frage gestellt. Der genetische Nachweis von HLA-DRB1*04 scheint ein starker Risikofaktor für die Entwicklung einer RZA zu sein (2). Bei der TA findet sich gehäuft die Konstellation HLA-B*52 (3). Möglicherweise erklärt diese genetische Differenz den unterschiedlichen Phänotyp der beiden Krankheiten bezüglich ethnischer als auch klinischer Manifestation bei praktisch gleicher Histologie der Gefässentzündung. Die PMR wirkt primär wie eine «rheumatische» Erkrankung von Synovia und gelenksnahen Strukturen (4). Allerdings teilen auch PMR und RZA einen genetischen Polymorphismus im HLA-DRB1 Gen (5). Rund 50% der RZA präsentieren sich auch als PMR, initial oder im Verlauf, resp. die PMR entwickelt sich zur RZA (6, 7). In 18FDG-PET-Untersuchungen finden sich bei 30-90 % der PMR-Patienten arteriitische Veränderungen der Wände von Aorta und der abgehenden grossen Gefässe (6, 8, 9). Es ist zu vermuten, dass die PMR eher Frühstadium resp. klinischer Ausdruck der Entzündungsmediatoren der Arteriitis denn ein eigenständiges Weichteil-Krankheitsbild ist. Dafür sprechen auch histologische Befunde, wo bei PMR aktivierte dendritische Zellen in der Adventitia, aber noch keine die Media infiltrierende T-Lymphozyten gefunden wurden (10). Zwar gibt es im klinischen Alltag durchaus noch die typischen Manifestationen der Polymyalgia rheumatica, der Arteriitis temporalis, beide bei älteren Patienten und die Takayasu-Arteriitis der jungen Frau. Die neuen Erkenntnisse und Bildgebungen aber auch unsere eigenen Beobachtungen legen indessen nahe, dass grosse Überlappungen, vor allem zwischen RZA und Polymyalgie bestehen, und die Takayasu-Arteriitis, wenn sie bei einem über 40-jährigen Patienten auftritt, durchaus Ähnlichkeiten mit einer RZA aufweist. Die Unterschiede liegen wahrscheinlich nicht in unterschiedlichen Ätiologien, sondern in einer anderen genetischen Disposition der befallenen Individuen und im Stadium, wo die Krankheit entdeckt wird.
Epidemiologie
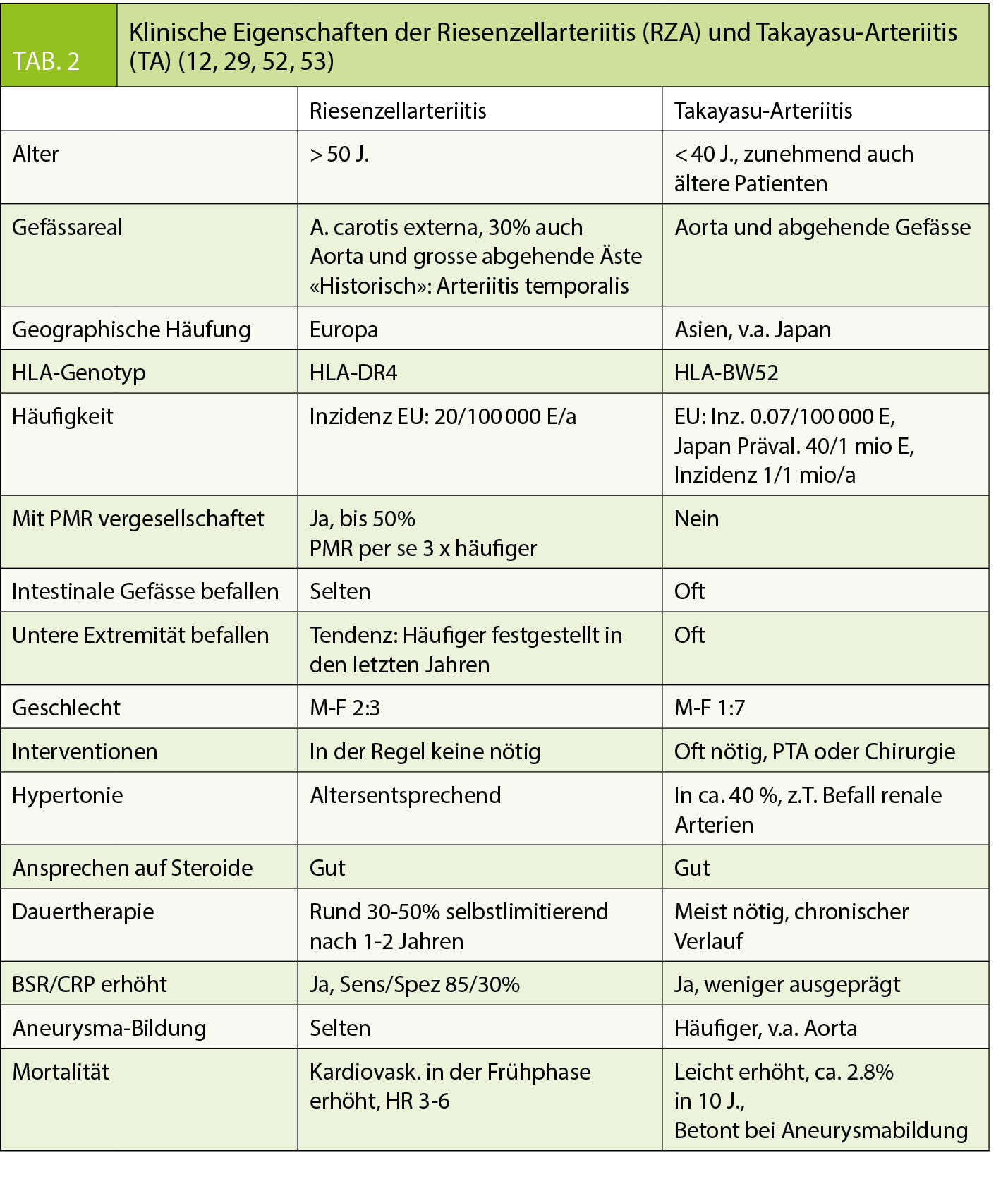
Für RZA und PMR liegt die Inzidenz in Europa bei ca. 20/100 000 E/a. Die TA ist vor allem in Japan bekannt, die bekannten Prävalenzzahlen liegen dort um 40/1 Mio. Einwohner. In der Regel sind RZA-Patienten über 50 Jahre alt, meist zwischen 70 und 80 und etwas häufiger weiblich. Bei der TA sind Frauen 7 x häufiger betroffen und meist unter 40-jährig (11, 12).
Symptomatik
Initial sind die Symptome unspezifisch und Ausdruck der massiven Ausschüttung von Entzündungsmediatoren. In späteren Stadien entsprechen sie der Schädigung von betroffenen Organen durch die entzündlich bedingten Gefässobstruktionen, z.B. die Erblindung bei Befall der Augenarterien. Eine Frühdiagnose von PMR, RZA oder TA auf Grund einer klinischen Beurteilung gilt auch heute noch als diagnostische Meisterleistung und bringt viel Ehre. Wichtig ist, bei allen unklaren Krankheitsbildern mit Müdigkeit, Schmerzen, Fieber und AZ-Verschlechterung an die Möglichkeit einer Arteriitis zu denken. Bei der PMR weisen beim Patienten über 50 Jahre die klassischen morgendlichen Schulter- oder Beckenschmerzen, bei der RZA die temporalen Kopfschmerzen oder die Kieferclaudicatio auf die Möglichkeit dieser Diagnosen. Bei der TA gibt es diese «klassische» Symptomatik nicht. Tab. 2 listet diagnostische Merkmale der 2 Entitäten auf. Besonders wichtig ist die rasche Diagnose bei der RZA mit Temporalarterienbefall. Wenn sich der Kopfschmerz atypisch-«nicht-temporal»- manifestiert, können diagnostische Irrwege mit der verheerenden Konsequenz einer Erblindung resultieren. Eine Amaurosis fugax kann erster und letzter Vorbote des drohenden Sehverlustes sein und bedarf in Kombination mit Kopfschmerzen und Allgemeinsymptomen einer notfallmässigen Diagnostik mit dem Ziel einer Steroidtherapie am gleichen Tag. Als erste Anlaufstelle ist beim Grundversorger ein «high index of suspicion» wichtigstes Prinzip der Primärdiagnostik.
Diagnostik
Klinische Untersuchungsbefunde
Die Palpation der Pulse ist essentiell. Sind sie abgeschwächt oder fehlend, hat die Arteriitis bereits zu einer hochgradigen Stenosierung oder gar zu einem Verschluss des Gefässes geführt. Die Pulsbeurteilung ist an der A. temporalis oft schwierig. Bei Arteriitis temporalis ist vor allem die Kombination von verdicktem und lokalisiert schmerzhaftem Gefässstrang eine brauchbare klinische Spur. Die Blutdruckmessung bestätigt Stenosen oder Verschlüsse semiquantitativ, wenn Seitendifferenzen von über 10 mm Hg vorliegen. Auch ungewöhnliche Gefässgeräusche, z.B. in der A. axillaris, können auf eine entzündliche Gefässstenosierung weisen.
Laboruntersuchungen
Einen spezifischen Laborparameter gibt es nicht. Bei den einfachen Grunduntersuchungen haben CRP (C-reaktives Protein) und BSR (Blutsenkungsreaktion) den höchsten Stellenwert. Sie erreichen bei der PMR resp. der RZA eine Sensitivität von fast 90% bei aber sehr geringer Spezifität von etwa 30%. Klassisch ist die sehr hohe BSR von > 100 mm, aber rund 4 % der Patienten haben normale BSR- und CRP-Werte (13). Bei der TA ist die Sensitivität etwas niedriger mit etwa 70%. Bis 25% der Patienten haben keine Erhöhung von CRP und BSR (14). Damit eignen sich beide Parameter in der Primärdiagnostik weder für den Beweis noch den Ausschluss von RZA und TA. Ihr Platz ist vor allem in der einfachen Monitorisierung des Krankheitsverlaufes.
Bildgebungen und Biopsie
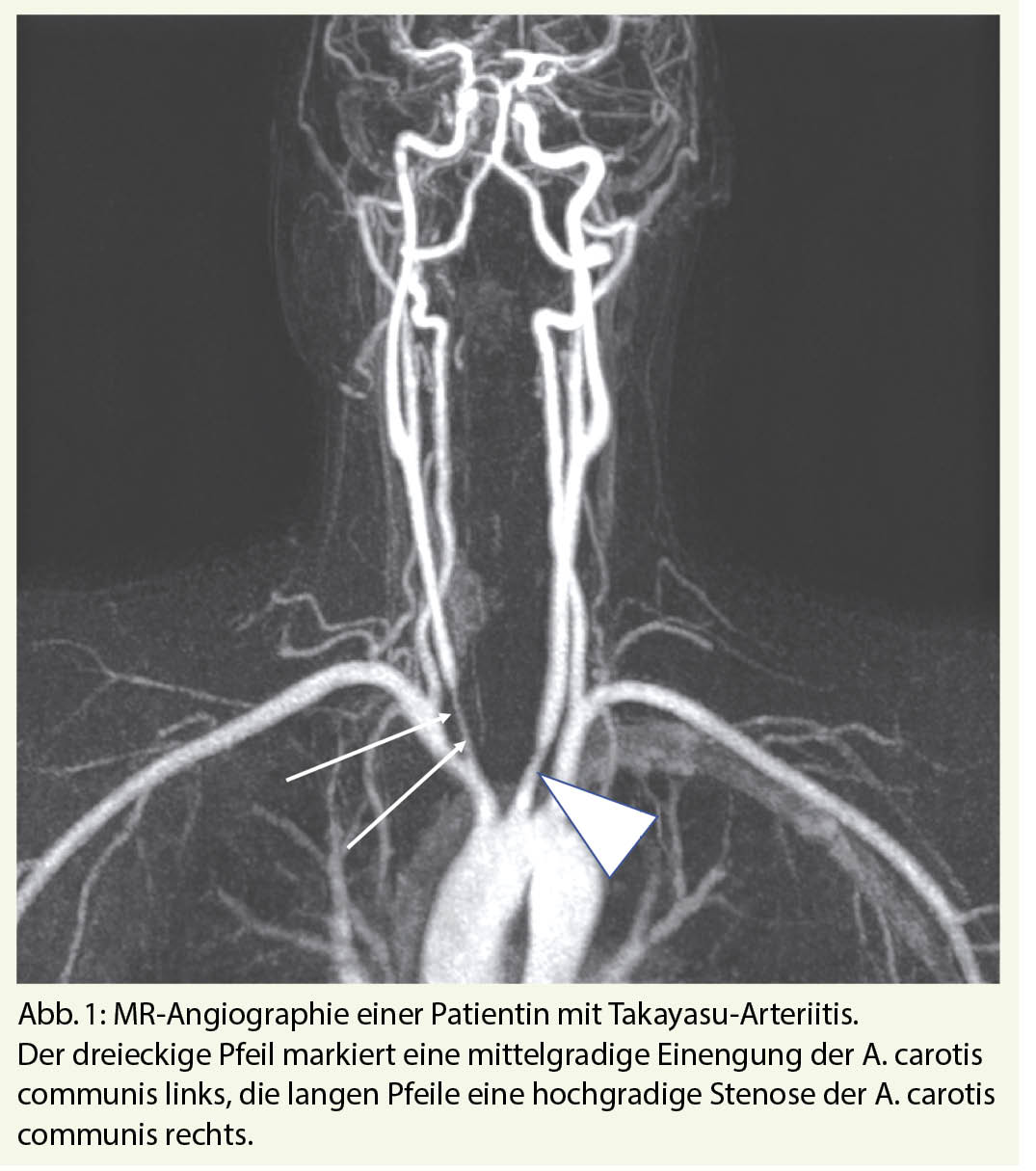
Computertomographie (CT), Magnetresonanztomographie (MR), 18FDG-Positronen-Emissions-Tomographie (PET) und die Duplex-Sonographie sind die wichtigen bildgebenden Verfahren. CT und MR haben den Vorteil der guten Übersichtsdarstellung mit simultaner Erfassung vieler oder aller befallenen Gefässabschnitte inklusive Aorta. In Abb. 1 ist der Befall der A. carotis communis bds. bei TA mittels MR-Angiographie dargestellt.
Die PET, auch kombiniert mit dem CT, kann die entzündliche Aktivität der Gefässwände sehr gut erfassen und erreicht bei einer Grossgefässvaskulitis eine Sensitivität und Spezifität von 91 resp. 89% (15, 16).
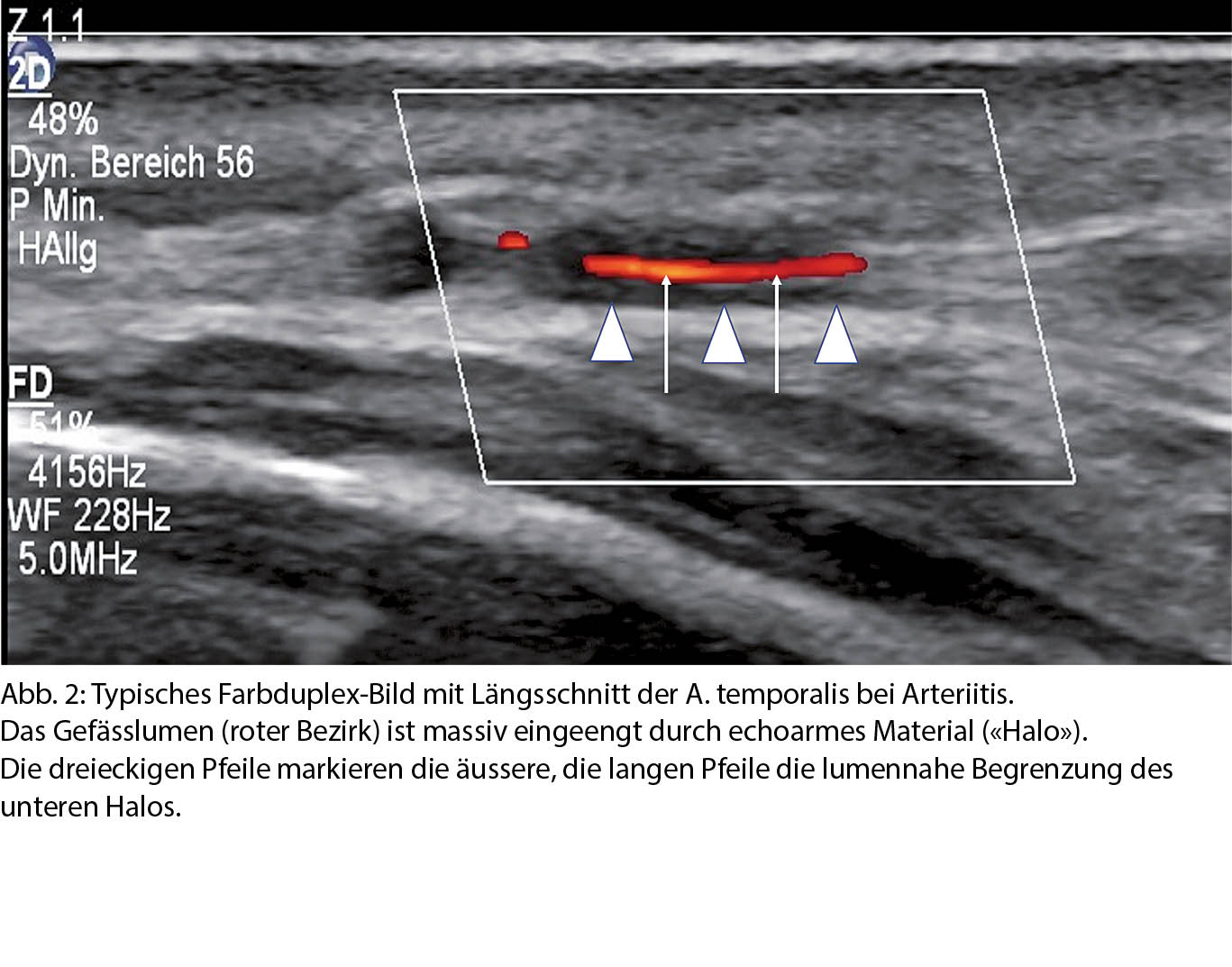
Besonders elegant ist die Duplex-Sonographie. Die nicht-invasive und kostengünstige Methode kann sowohl Wandveränderungen als auch Blutflusscharakteristika darstellen und liefert über den arteriitischen Prozess morphologische und hämodynamische Informationen. (17, 18). Hypoechogene Wandpolster, sog. Halo, die das Lumen in unterschiedlichem Mass einengen können, sind typisch für die RZA. Die Sensitivität und Spezifität für die Diagnose der RZA der Duplex-Sonographie beträgt 69 resp. 91%, vergleichbar mit der MRT (19). Damit kann die Krankheit bei unauffälliger Duplexsonographie zwar nicht ausgeschlossen werden, wenn aber sonographisch arteriitische Befunde nachweisbar sind, ist die Beweiskraft hoch. Ein positiver Ultraschallbefund korreliert mit dem klinischen Verlauf einer RZA nach einer neueren Untersuchung sogar besser als die Biopsie der A. temporalis (20). Typische Klinik, passende Entzündungsparameter im Labor und eine arteriitische Wandveränderung in der Ultraschalluntersuchung sichern die Diagnose einer RZA oder einer Polymyalgie mit Riesenzell-
arteriitis und machen in der Regel eine Biopsie überflüssig (21). Abb. 2 zeigt einen typischen sonographischen Befund einer floriden RZA der Temporalarterie.
Bei der TA sind die Wandauflagerungen der etwas grösseren Gefässe in der Regel etwas echodichter (22). Auch hier kann der Prozess eine Lumeneinengung verursachen, die mit der Duplexsonographie quantifiziert werden kann. Die Sensitivität und Spezifität für die Diagnose von Stenosen bei TA beträgt 90 resp. 91% (23). Kontrastmittelverstärkter Ultraschall und spezielles Scoring der Wandveränderungen inkl. IMT sind weitere diagnostische Möglichkeiten deren abschliessende Beurteilung aussteht (24, 25).
Differentialdiagnosen
Das unspezifische Frühstadium einer Grossgefässvaskulitis kann viele Krankheiten durch Ausschüttung von Zytokinen und anderen Entzündungsmediatoren nachahmen, z.B. Infekte, Tumore, Sarkoidose, rheumatische und andere Autoimmunerkrankungen (11). Die Abgrenzung ist insbesondere bei der PMR schwierig. Ist die Wandentzündung fortgeschritten, wird die Spur, z.B. bezüglich RZA mit Temporalarterienbefall oder Subclavia-Stenose bei TA, konkreter. Im letzteren Fall ist die Abgrenzung gegen die häufige arteriosklerotisch bedingte PAVK wichtig. Alter, vorbestehende Risikofaktoren und bereits durchgemachte vaskuläre Zwischenfälle helfen bei der Unterscheidung.
Therapeutische Aspekte
Nach wie vor sind Steroide die Eckpfeiler der Behandlung von Arteriitiden (13, 26, 27, 28, 29, 30). Dabei werden 3 Régimes angewendet:
1. Hohe intravenöse Stosstherapie bei RZA mit Augensymptomen oder bereits eingetretenem Visusverlust. Hier werden 3 Tage 500-1000 mg Methylprednisolon verabreicht, dann Prednison 1 mg/kg KG pro Tag, max. 60 mg, peroral, dann Reduktion der Dosis um 10-20% pro 2 Wochen, nach Erreichen von 10 mg/Tag weitere Reduktion um 1 mg /Monat. Parallel zur Reduktion der Steroide klinische Kontrolle und monatliche Kontrollen der Entzündungswerte im ersten Jahr, alle zwei Monate im zweiten Jahr und dann alle 3-6 Monate.
2. RZA ohne Augensymptome und TA werden mit peroralem Prednison, 1 mg/kg KG/Tag behandelt, Dosisreduktion gemäss obenstehender Empfehlung.
3. Die Polymyalgia rheumatica ohne Gefässbefall lässt sich gut mit niedrigeren Dosen behandeln, in der Regel 15-20 mg Prednison/Tag während 2-4 Wochen, anschliessend 10 Wochen 10-15 mg und dann ebenfalls mg-weise Reduktion jeden Monat unter Kontrolle von Klinik, BSR und Labor.
RZA und PMR können selbstlimitierend sein. Ein Absetzversuch der Steroide nach 1-2 Jahren ist deshalb sinnvoll. Bei 30-70% der Patienten kommt es allerdings zu einem Wiederaufflackern der Symptomatik (31, 32). Bei der TA ist der Verlauf in der Regel chronisch und ein vollständiges Absetzen der Medikation nicht möglich. Über die Hälfte der Patienten mit Arteriitis braucht wegen anhaltend hoher Steroid-Dosen eine medikamentöse Alternative (33, 34).
Am häufigsten angewendet wird Methotrexat (35, 36). Azathioprin und Cyclophosphamid sind andere Alternativen (37, 38). Tocilizumab, ein Interleukin-6-Antagonist, ist das bestuntersuchte Biological bei RZA und TA und spart bei vertretbarem Nebenwirkungsprofil fast die Hälfte der Steroiddosis (39). Für TA sind auch Leflunomid und Mycophenolat in kleinen Serien untersucht (40, 41). TNF-Inhibitoren scheinen im Gegensatz zur Anwendung bei RZA eine gewisse Wirkung zu haben, bei allerdings erheblichen Nebenwirkungen (42). Die Indikation zu diesen teuren und/oder nebenwirkungsträchtigen Therapien wird in der Regel von Spezialisten mit entsprechender Erfahrung gestellt.
Diverse flankierende Massnahmen sind zu beachten. Alle Patienten mit einer geplanten Steroid-Therapie über 3 Monate müssen bezüglich Osteoporose-Risiko gescreent werden und minimal Vitamin D und Calcium bekommen. Je nach Risikokonstellation sind eine antiresorptive Therapie und entsprechende Kontrollen indiziert (43). Aspirin hat keinen Effekt in der Behandlung der Arteriitis, soll aber bei Indikation aus anderen Gründen weitergeführt werden (44, 45). Eine Prophylaxe gegen Pneumocystis jirovecii wird bei Kombinationstherapie Steroide-Methotrexat empfohlen (46, 47).
Währenddem bei RZA die Immunsuppression ausreicht, können bei TA Interventionen nötig sein. Kurzstreckige Stenosen lassen sich mit Angioplastie behandeln, die Rolle des zusätzlichen Stentings ist offen (48, 49). Bei langen, fibrotisch veränderten Segmenten ist die Bypass-Operation in der chronischen Krankheitsphase bevorzugte Methode. Bei Notwendigkeit von Überbrückungen nach brachio-kranial oder koronar sollte der proximale Anschluss wenn immer möglich in der Aorta ascendens liegen, da weiter peripher liegende Arterien häufig in den Krankheitsprozess miteinbezogen sind (50, 51).
Hausarzt oder Spezialist oder beide?
Eine typische Polymyalgie kann durch einen engagierten Grundversorger behandelt werden. Wichtig ist die rasche Reduktion der Steroide auf die minimal nötige Erhaltungsdosis und der Absetzversuch nach 1-1.5 Jahren. Zudem gehört die Abschätzung des Osteoporoserisikos zum obligatorischen Programm. Sobald Probleme auftreten, z.B. ungenügendes Ansprechen, erhebliche Steroid-Nebenwirkungen oder bei Osteoporose oder -penie, soll der Patient dem Spezialisten überwiesen werden.
Wegen der schwierigeren Diagnostik und der potentiellen Komplikationen gehören TA, RZA und die PMR mit Gefässbefall in Spezialistenhände. Nach festgelegtem Therapieplan sind diese Patienten mit einer integrierten Versorgung Hausarzt-Spezialist-Zentrum in den besten Händen.
Dr. med. Alexander von Weymarn
Gefässzentrum USGG Spital Thurgau Frauenfeld
Interventionelle Radiologie Institut für Radiologie
Kantonsspital
8500 Frauenfeld
Dr. med. Saulius Sudikas
Gefässzentrum USGG Spital Thurgau Frauenfeld
Gefässchirurgie Chirurgische Klinik
Kantonsspital
8500 Frauenfeld
Dr. med. Beat Bundi
Gefässzentrum USGG Spital Thurgau Frauenfeld
Angiologie Medizinische Klinik,
Kantonsspital
8500 Frauenfeld
Gefässzentrum USGG Spital Thurgau Frauenfeld
Angiologie Medizinische Klinik,
Kantonsspital, 8500 Frauenfeld
beat.frauchiger@stgag.ch
Die Autoren haben in Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
1. Jennette JC et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65(1):1-11
2. Carmona FD et al. A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am J Hum Genet 2015;96(4):565-80
3. Renauer P et al. The genetics of Takayasu arteritis. Press Med 2017; 46: e179-e187
4. Chuang TY et al. Polymyalgia rheumatica: a 10-year epidemiologic and clinical study. Ann Intern Med 1982;97(5):672-80
5. Weyand CM et al. HLA-DRB1 alleles in polymyalgia rheumatica, giant cell arteritis, and rheumatoid arthritis. Arthritis Rheum 1994;37(4):514-20
6. Dejaco C et al. The spectrum of giant cell arteritis and polymyalgia rheumatica: revisiting the concept of the disease. Rheumatology 2017;56(4):506-15
7. Cantini F et al. Are polymyalgia rheumatica and giant cell arteritis the same disease? Semin Arthritis Rheum 2004;33(5):294-301
8. Moosig F et al. Correlation between 18-fluorodeoxyglucose accumulation in large vessels and serological markers of inflammation in polymyalgia rheumatica: a quantitative PET study. Ann Rheum Dis 2004;63(7):870-73
9. Blockmans D et al. Repetitive 18-fluorodeoxyglucose positron emission tomography in isolated polymyalgia rheumatica: a prospective study in 35 patients. Rheumatology 2007;46(4):672-77
10. Weyand CM et al. Medium- and large-vessel vasculitis. N Engl J Med 2003; 349(2):160-9
11. Watts RA et al. Introduction, epidemiology and classification of vasculitis. Best Pract Res Clin Rheumatol 2018;32(1):3-20
12. Alibaz-Öner F et al. Recent advances in Takayasu’s arteritis. Eur J Rheumatol 2015;2(1):24-30
13. Kermani TA et al. Polymyalgia rheumatica. Lancet 2013;5;381:63-72
14. Kerr GS et al. Takayasu arteritis. Ann Intern Med 1994;1;120(11):919-29
15. Andrews J et al. Non-invasive imaging in the diagnosis and management of Takayasu’s arteritis. Ann Rheum Dis 2004;63(8):995-1000
16. Fuchs M et al. The impact of 18F-FDG PET on the management of patients with suspected large vessel vasculitis. Eur J Nucl Med Mol Imaging 2012; 39(2):344-53
17. Schmidt WA et al. Ultrasound of proximal upper extremity arteries to increase the diagnostic yield in large-vessel giant cell arteritis. Rheumatology 2008;47(1):96-101
18. Schmidt WA et al. Do temporal artery duplex ultrasound findings correlate with ophthalmic complications in giant cell arteritis? Rheumatology 2009;48(4):383-5
19. Bley TA et al. Comparison of duplex sonography and high-resolution magnetic resonance imaging in the diagnosis of giant cell (temporal) arteritis. Arthritis Rheum 2008;58(8):2574-8
20. Mukhtyar C et al. Validating a diagnostic GCA ultrasonography service against temporal artery biopsy and long-term clinical outcomes. Clin Rheumatol 2019; 10.1007/s10067-019-04772-2, Epub
21. Schmidt WA. Ultrasound in the diagnosis and management of giant cell arteritis. Rheumatology 2018;1;57(suppl_2):ii22-ii31
22. Nicoletti G et al. The “Macaroni Sign” of Takayasu’s arteritis. J Rheumatol 2009;36(9):2042-3
23. Raninen RO et al. Ultrasonography in the quantification of arterial involvement in Takayasu’s arteritis. Scand J Rheumatol 2000;29(1):56-61
24. Schinkel AF et al. Utility of contrast-enhanced ultrasound for the assessment of the carotid artery wall in patients with Takayasu or giant cell arteritis. Eur Heart J Cardiovasc Imaging 2014;15(5):541-6
25. Sinha et al. Development of a colour Doppler ultrasound scoring system in patients of Takayasu’s arteritis and its correlation with clinical activity score (ITAS) 2010. Rheumatology 2013;52(12):2196-202
26. Dasgupta B et al. BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology 2010;49(8):1594-7
27. Mukhtyar C et al. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2009;68(3):318-23
28. Delecoeuillerie G Pet al. Polymyalgia rheumatica and temporal arteritis: a retrospective analysis of prognostic features and different corticosteroid regimens (11 year survey of 210 patients). Ann Rheum Dis 1988;47(9):733-9
29. Weyand CM et al. Giant-cell arteritis and polymyalgia rheumatica. N Engl J Med 2014;3;371(1)50-7
30. Keser G et al. Management of Takayasu arteritis: a systematic review. Rheumatology 2014;53(5):793-801
31. Kermani TA et al. Disease Relapses among Patients with Giant Cell Arteritis: A Prospective, Longitudinal Cohort Study. J Rheumatol 2015;42(7):1213-7
32. Labarca C et al. Predictors of relapse and treatment outcomes in biopsy-proven giant cell arteritis: a retrospective cohort study. Rheumatology 2016;55(2):347-56
33. Hoffman GS el al. Treatment of glucocorticoid-resistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum 1994;37(4):578-82
34. Mekinian A et al. Efficacy of Biological-Targeted Treatments in Takayasu Arteritis: Multicenter, Retrospective Study of 49 Patients. Circulation 2015;132(18):1693-700
35. Jover JA et al. Combined treatment of giant-cell arteritis with methotrexate and prednisone. A randomized, double-blind, placebo-controlled trial. Ann Intern Med 2001;134(2):106-14
36. Spiera RF et al. Methotrexate in giant-cell arteritis. Ann Intern Med. 2001;135(11):1006-7
37. De Silva M et al. Azathioprine in giant cell arteritis/polymyalgia rheumatica: a double-blind study. Ann Rheum Dis 1986;45(2):136-8
38. Quartuccio L et al. Role of oral cyclophosphamide in the treatment of giant cell arteritis. Rheumatology 2012;51(9):1677-86
39. Villiger PM et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016;387(10031):1921-7
40. de Souza AW et al. Leflunomide in Takayasu arteritis – A long term observational study. Rev Bras Reumatol Engl Ed. 2016;56(4):371-5
41. Daina E et al. Mycophenolate mofetil for the treatment of Takayasu arteritis: report of three cases. Ann Intern Med 1999;130(5):422-6
42. Barra L et al. Non-glucocorticoid drugs for the treatment of Takayasu’s arteritis: A systematic review and meta-analysis. Autoimmun Rev 2018;17(7):683-693
43. Buckley L et al. 2017 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis. Arthritis Rheumatol 2017;69(8):1521-1537
44. Lee MS et al. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum 2006;54(10):3306-9
45. Salvarani C et al. Risk factors for severe cranial ischaemic events in an Italian population-based cohort of patients with giant cell arteritis. Rheumatology 2009;48(3):250-3
46. Kermani TA et al. Pneumocystis jiroveci pneumonia in giant cell arteritis: A case series. Arthritis Care Res 2011;63(5):761-5
47. Berger CT et al. Risk factors for pneumocystis pneumonia in giant cell arteritis: a single-centre cohort study. Clin Exp Rheumatol 2015;33(2 Suppl 89):S-122-5
48. Ogino H, et al. Overview of late outcome of medical and surgical treatment for Takayasu arteritis. Circulation 2008;118(25):2738-47
49. Jeong HS, et al. Endovascular balloon angioplasty versus stenting in patients with Takayasu arteritis: A meta-analysis. Medicine (Baltimore). 2017;96(29):e7558
50. Serra R, et al. Updates in Pathophysiology, Diagnosis and Management of Takayasu Arteritis. Ann Vasc Surg 2016;35:210-25
51. Mason JC. Surgical intervention and its role in Takayasu arteritis. Best Pract Res Clin Rheumatol 2018;32(1):112-24
52. Michel BA et al. Clinical differentiation between giant cell (temporal) arteritis and Takayasu’s arteritis. J Rheumatol 1996;23(1):106-11.
53. Furuta S et al. Clinical features and radiological findings in large vessel vasculitis: are Takayasu arteritis and giant cell arteritis 2 different diseases or a single entity? J Rheumatol 2015;42(2):300-8.

der informierte @rzt
- Vol. 9
- Ausgabe 12
- Dezember 2019