
Kollagenosen sind eine Gruppe seltener, Autoimmuner Erkrankungen, welche durch ihr zum Teil sehr heterogenes klinisches Erscheinungsbild insbesondere bei Krankheitsbeginn schwierig zu diagnostizieren sind. Hinzu kommt, dass die Krankheitsbilder sich teilweise überlappen und zu Beginn ein unspezifisches klinisches Bild zeigen können. Im folgenden Artikel wollen wir einen Überblick über die Krankheitsbilder geben und mögliche Abklärungsschritte aufzeigen. Wir verzichten bewusst auf eine Diskussion der Therapie, da dies den Rahmen dieses Artikels sprengen würde.
Collagenoses are a group of rare, autoimmune diseases, which are difficult to diagnose due to their partly very heterogeneous clinical appearance, especially at the onset of the disease. In addition, the disease patterns partly overlap and may show a non-specific clinical picture at the beginning. In the following article we would like to give an overview of the clinical pictures and point out possible diagnostic approaches. We deliberately refrain from discussing therapy, as this would go beyond the scope of this article.
Key Words: collagenoses, autoimmune diseases
Was sind Kollagenosen?
Der Begriff Kollagenosen scheint irreführend, da im Gegensatz zu den Kollagenkrankheiten wie der Osteogenesis-Imperfecta bei den Kollagenosen keine Störung des Kollagens besteht. Vielmehr handelt es sich um systemische, entzündliche, autoimmune Erkrankungen, welche das Bindegewebe aber auch verschiedene Organe betreffen können. Entsprechend wird im englischen Sprachgebrauch auch der Begriff der systemisch entzündlichen Bindegewebserkrankungen (Connective tissue diseases, kurz CTD) verwendet. Kollagenosen treten häufiger bei jungen Frauen auf, können aber grundsätzlich Frauen und Männer jeden Alters (und auch Kinder) betreffen.
Zu den häufigsten Kollagenosen gehören das Sjögren-Syndrom, der systemische Lupus erythematodes (SLE) und die systemische Sklerose (Sklerodermie). Die rheumatoide Arthritis (in diesem Artikel nicht vertieft behandelt) wird oft auch den Kollagenosen zugeordnet, ist jedoch durch den prominenten Gelenkbefall und der meistens weniger ausgeprägten extra-artikulären Symptomatik von den anderen abgesetzt. Seltenere Kollagenosen sind die idiopathischen inflammatorischen Myopathien (Myositiden) und die Mischkollagenose (MCTD: mixed connective tissue disease), sowie noch seltenere Erkrankungen wie die relapsing Polychondritis auf welche wir in diesem Artikel nicht weiter eingehen werden. Folgend werden wir die häufigsten Kollagenosen kurz beschreiben und dann auf eine Kollagenose hinweisende Symptome besser beleuchten.
Kollagenose-Diagnostik, Klassifikationskriterien, undifferenzierte Kollagenosen, Overlap-Syndrom und Mischkollagenose
Die Diagnose der Kollagenosen basiert immer auf der Kombination der gründlichen Anamnese, der klinischen Untersuchung und der laboranalytischen Befunde, situativ ergänzt mit Befunden aus Bildgebung und Histologie (z.B. Nierenbiopsie).
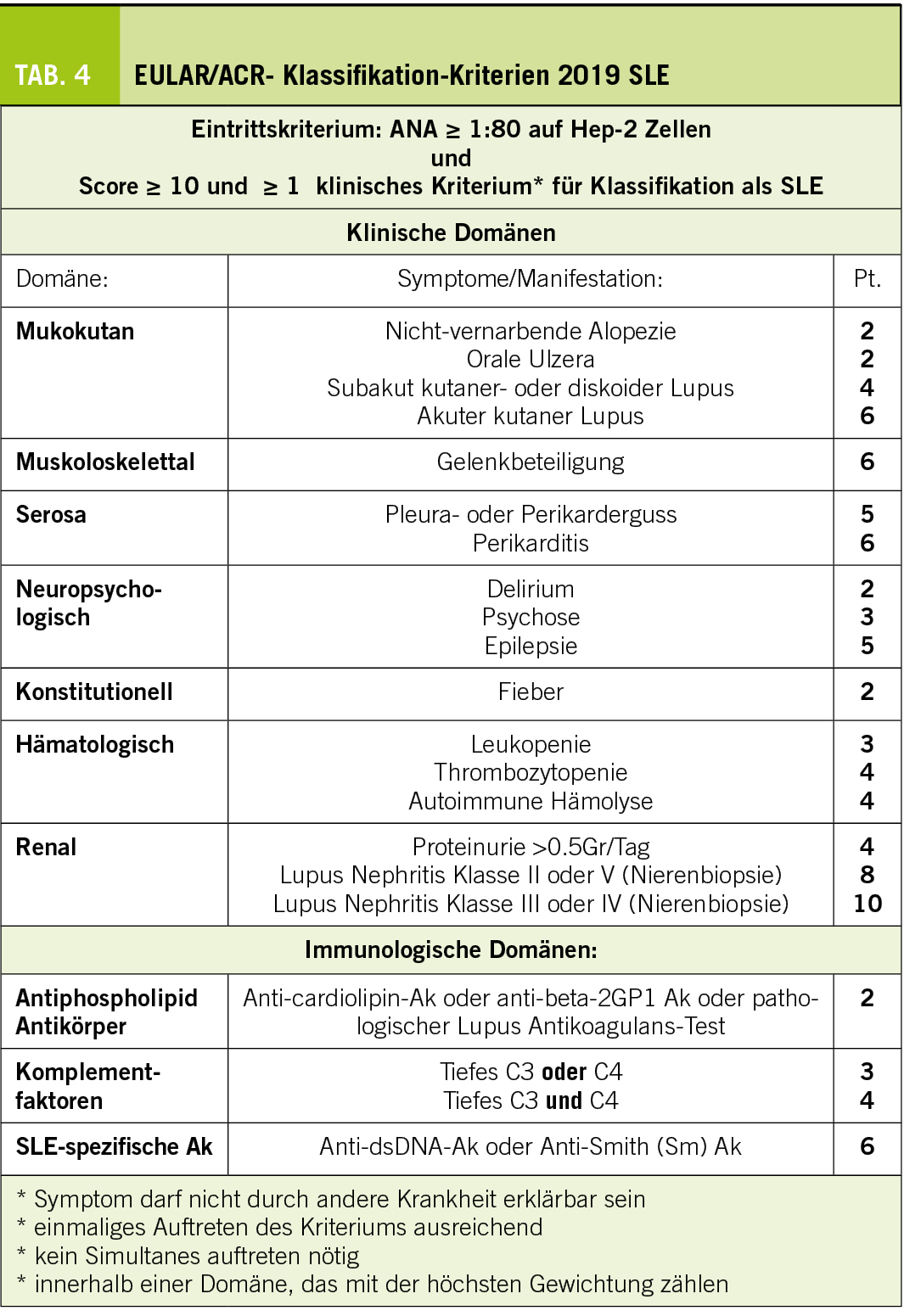
Da keine Diagnosekriterien existieren, ist der Goldstandard für die Diagnose der verschiedenen Kollagenosen die «Expert:Innen Meinung». Entsprechend unscharf ist die Abgrenzung der einzelnen Erkrankungen gegeneinander, aber auch gegenüber anderen systemischen entzündlichen Erkrankungen. Zwar existieren für wissenschaftliche Zwecke entwickelte Klassifikationskriterien (Tab. 4), damit innerhalb von Studien ein möglichst homogenes Kollektiv an Patien:Innen gefunden wird, diese sind aber nicht zur Diagnose gedacht (auch wenn sie hierfür oft herangezogen werden). Es muss bewusst sein, dass nicht rein aufgrund dieser Klassifikationskriterien die Diagnose gestellt oder noch wichtiger verworfen wird. Zum Beispiel wäre es falsch einen SLE zu diagnostizieren nur weil eine Patient:In Arthralgien, Fieber und positive anti-ds-DNA-Antikörper aufweist, ohne zuerst einen Infekt auszuschliessen. Auch ist es falsch bei einer Patientin mit positiven anti-Zentromer-Antikörpern, puffy fingers und Raynaud-Phänomen eine frühe systemische Sklerose nicht zu diagnostizieren, weil sie «nur» 8 von 9 Klassifikationskriterien gemäss den 2013-Kriterien für Systemische Sklerose erfüllt.
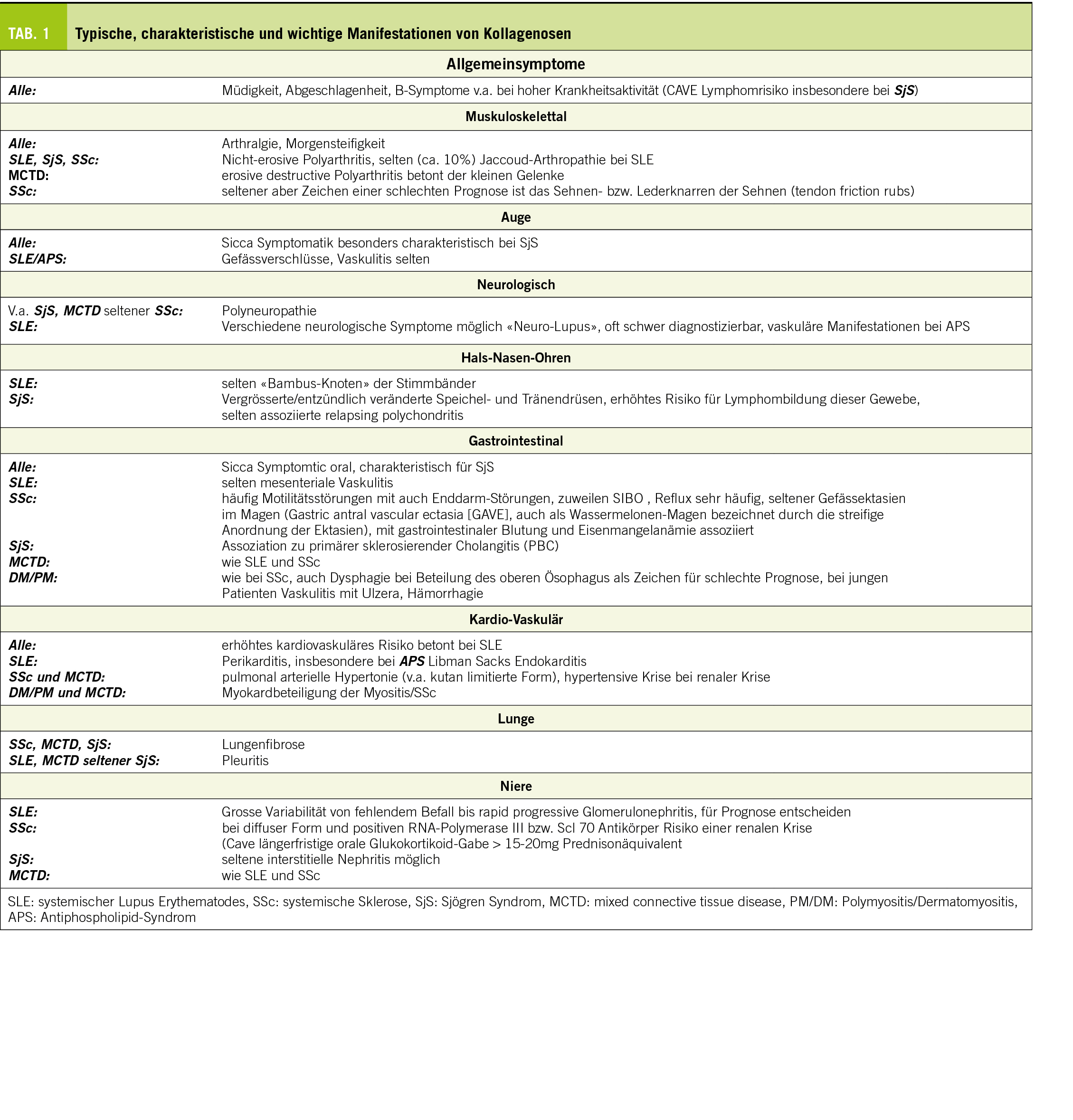
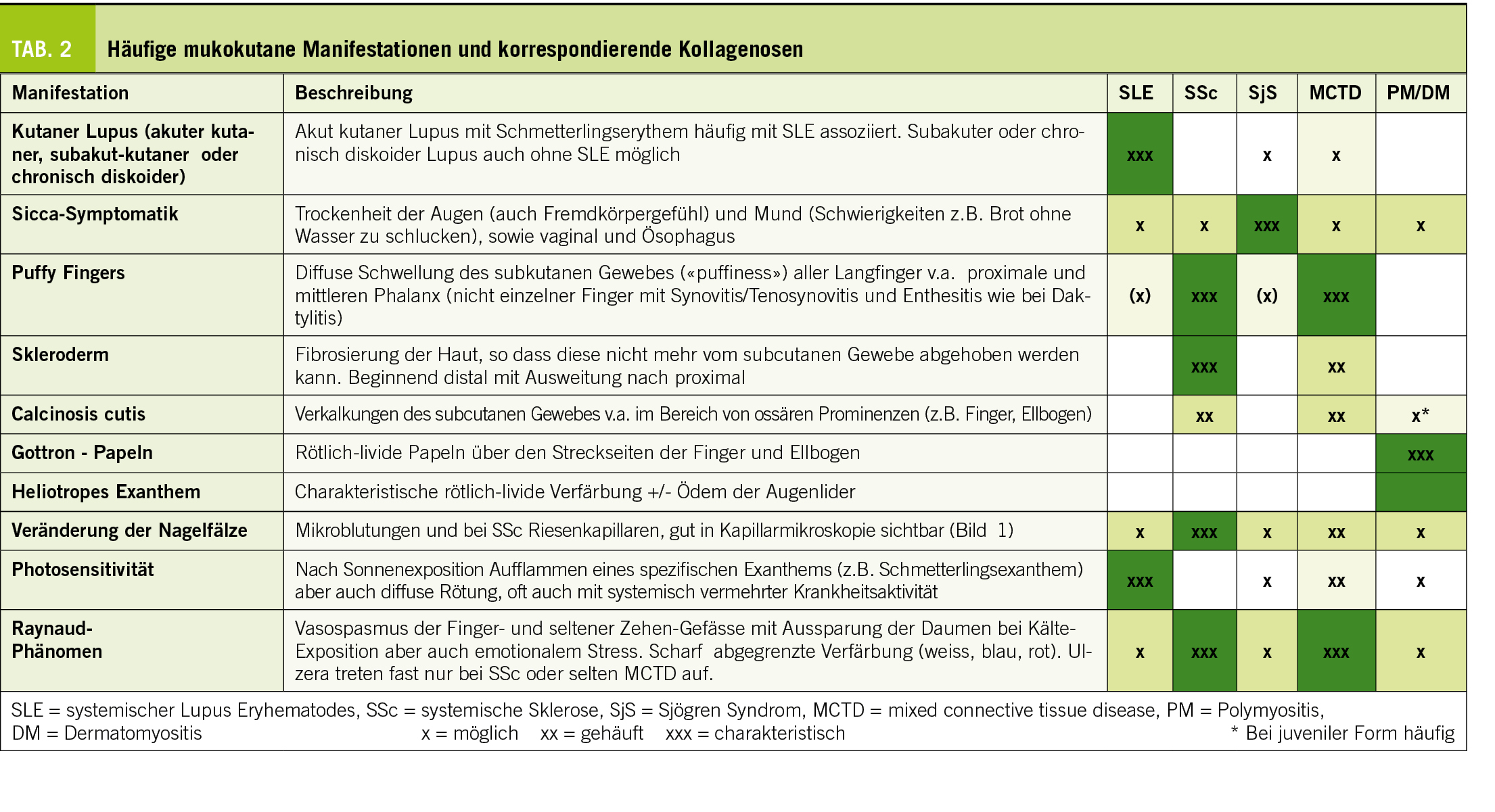
Eine Übersicht zu typischen Symptomen findet sich in Tabelle 1 sowie gesondert der mukokutanen Manifestation in Tabelle 2 und Labor bei Tabelle 5.
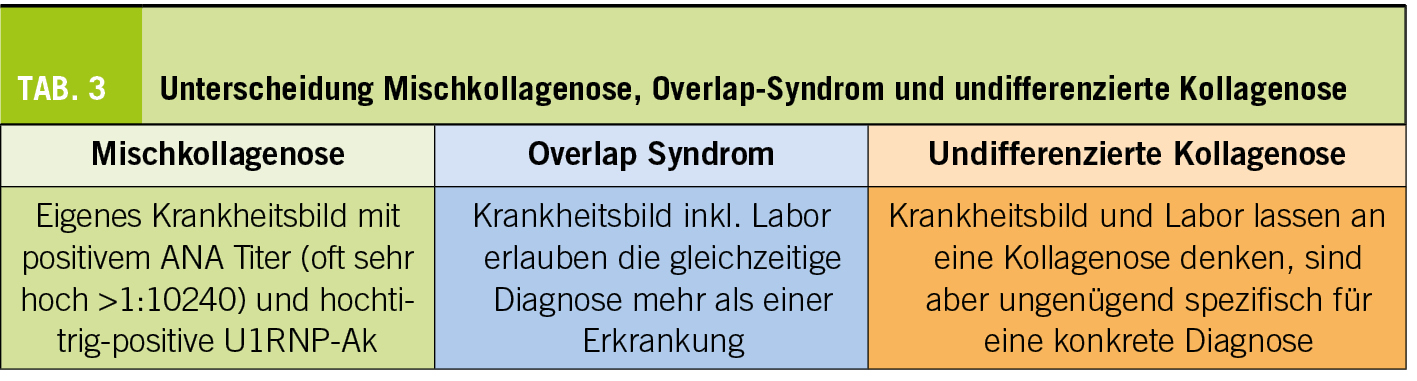
Nicht selten zeigt sich aber, insbesondere zu Beginn der Erkrankung, nicht das Vollbild einer Erkrankung oder es bleibt auch im weiteren Verlauf bei Manifestationen, welche keine eindeutige Kollagenose diagnostizieren lassen (z.B. Patientin mit Raynaud-Phänomen, Photosensitivität und erhöhten Antinukleären Antikörpern ohne spezifische Antikörper). In diesem Fall sprincht man von einer undifferenzierten Kollagenose. In diesen Fällen gilt es wachsam zu bleiben, damit man allenfalls im Verlauf eine konkretere Diagnose stellen kann, was aber nicht immer möglich ist.
Klar von einer undifferenzierten Kollagenose abzugrenzen ist die Mischkollagenose (mixed connective tissue disease, MCTD, auch Sharp – Syndrom genannt). Hierbei handelt es sich um eine klar umschriebene Erkrankung (siehe unten), welche Phänomene anderer Kollagenosen aufweist, die aber nicht diesen entspricht. Es handelt sich also nicht um eine Mischung von Kollagenosen. Liegen effektiv Befunde vor, welche die Diagnose mehrerer Kollagnosen zulassen spricht man von einem Overlap Syndrom. So würde man bei einer erosiven Arthritis mit positiven anti-CCP Antikörpern mit zusätzlich ds-DNA-Antikörpern und akutem kutanem Lupus von einem Overlap zwischen rheumatoider Arthritis und SLE sprechen, was zuweilen auch als «Rhupus» bezeichnet wird (Tab. 3).
Systemischer Lupus erythematodes
Der systemische Lupus erythematodes (SLE) ist das Rollenmodell einer Kollagenose. Er tritt gehäuft bei jungen Frauen auf und kann fast alle Organsysteme betreffen. Der typische Hautbefall im Sinne eines akuten kutanen Lupus erythematodes mit dem Schmetterlingserythem ist wohl den meisten bekannt. Hier muss es von einer Rosacea differenziert werden. Daneben besteht oft eine nicht nur auf die Haut bezogene Photosensitivität (Krankheitsschub nach vermehrter Sonnenexposition) sowie andere Formen des kutanen Lupus (z.B. subakuter kutaner Lupus) und Ulzera im Mund-, Nasen- und Rachenraum. Daneben bestehen oft Arthralgien und zuweilen auch veritable Arthritiden. Im Gegensatz zur rheumatoiden Arthritis sind Erosionen seltener, dafür kommt es zu Destruktionen des Kapsel-Band-Apparates mit charakteristischer Ulnardeviation der Finger, die sogenannte Jaccoud-Arthropathie. Gefürchtet ist der Nierenbefall, welcher die Krankheitsprognose entscheidend beeinträchtigt. Serositiden sind wie auch neuropsychiatrische Manifestationen selten, zuweilen aber für die Betroffenen schwer beeinträchtigend. Zusätzlich kann ein Antiphospholipid-Syndrom (APS) bestehen, was sowohl zu venösen als auch arteriellen Gefässverschlüssen führen kann. Bei arteriellen Thrombosen bei jungen Patienten und rezidivierenden venösen Thrombosen muss an ein APS gedacht werden. Aus allgemeinmedizinischer Sicht von grosser Relevanz ist das gesteigerte kardiovaskuläre Risiko, welches in dieser Population (junge Frauen) üblicherweise nicht vorhanden ist, entsprechend oft ungenügend beachtet wird, obwohl es für die Langzeitprognose der Patient:Innen von entscheidender Bedeutung ist, vor allem in Bezug auf die Mortalität und entsprechend streng kontrolliert und angegangen werden muss. Hier ist die Zusammenarbeit zwischen Spezialist:Innen und Allgemeinmediziner:Innen entscheidend.
Im Labor bestehen fast immer positive Antinukleäre Antikörper (ANA), wobei sich hier die Rate negativer Ergebnisse seit der zunehmenden Vereinheitlichung der Bestimmung der ANA verkleinert hat. Spezifische Antikörper sind diejenigen gegen Doppelstrang-DNA (auch als Verlaufsparameter hilfreich), Smith-Antikörper (Sm) und oft auch diejenigen des Antiphospholipid-Syndroms. Die Blutsenkungsgeschwindigkeit (BSG) ist oft erhöht, wohingegen das CRP nur selten und in spezifischen Fällen oder bei zusätzlicher Infektion erhöht ist. Daneben kann bei erhöhter Aktivität ein Komplementverbrauch bestehen, als Screening für Nierenbefall wird in der Urinanalyse nach aktivem Sediment und Proteinurie gesucht.
Sjögren Syndrom
Das Sjögren-Syndrom (SjS) betrifft ebenfalls häufiger Frauen, zeichnet sich klinisch primär durch den Befall der Speichel- und Tränendrüsen aus, was sich mit einer ausgeprägten Sicca-Symptomatik manifestiert. In diesem Zusammenhang kann auch eine generelle Trockenheit der Haut aber auch genital mit entsprechenden Beschwerden und Leidensdruck vorhanden sein. Daneben besteht oft Polyarthralgie (Polyar thritis selten) und Fatigue. Aufgrund dieses auch sonst häufigen Beschwerdebildes (Sicca-Symptomatik, Müdigkeit, Arthralgien), ist die Abgrenzung zu anderen Krankheitsbildern wie dem Diabetes mellitus, einer chronischen Hepatitis C und dem Fibromyalgie-Syndrom wichtig aber nicht immer ganz einfach (insbesondere letzteres kann auch parallel bestehen). Im Labor sind auch hier die ANA oft positiv, wobei auch ein erhöhter Rheumafaktor und spezifischere Antikörper gegen SS-A (auch als Ro bezeichnet) und SS-B (auch als LA bezeichnet) beobachtet werden. Die Laborbefunde sind aber nicht pathognomisch. Ergänzend kann eine Biopsie der kleinen Speicheldrüsen der Lippe erfolgen. Aus allgemeinmedizinscher- und prognostischer Sicht sollte das erhöhte Risiko für Lymphome beachtet und kontrolliert werden.
Systemische Sklerose («Sklerodermie»)
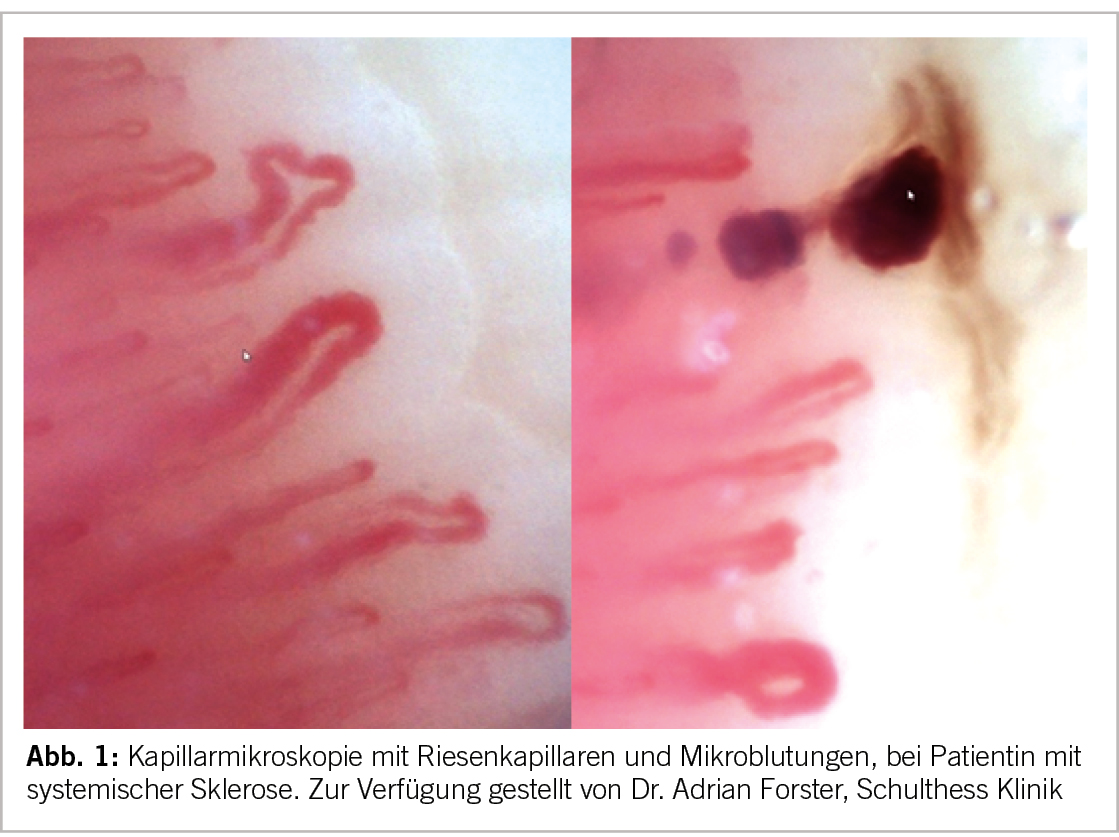
Namensgebend für diese, ebenfalls bei Frauen öfter auftretenden Erkrankung ist die Fibrosierung der Haut, welche zum sogenannten Skleroderm, also der hart werdenden Haut führt. Diese beginnt eigentlich immer an den Akren und im Gesicht und kann sich bei gewissen Patienten ausdehnen. Anhand des Hautbefalls wird die kutan limitierte (Skleroderm nach proximal bis maximal Knie- bzw. Ellbogenhöhe begrenzt, Gesicht aber möglich) und die kutan diffuse (Ausbreitung nach proximal über Knie- und Ellbogen hinaus) unterschieden. Diese Formen unterscheiden sich auch im Hinblick auf ihr Antikörperprofil, Organbeteiligung und damit auch Prognose. Es existieren auch Verläufe ohne Hautfibrose. Bei der kutan diffusen Form ist das Voranschreiten der Erkrankung oft aggressiver und v.a. die Lungenfibrose, sowie eine mögliche renale Krise (cave mit längerfristigen oralen Glucocorticoiden ≥ 15-20mg Prednison-Äquivalent) stehen im Fokus, wohingegen bei der kutan limitierten Form die pulmonal-arterielle Hypertonie (PAH) beachtet werden muss. Ein sehr häufiges Symptom und oft auch das Erstsymptom der systemischen Sklerose ist das Raynaud-Phänomen, weshalb bei einem Raynaud Phänomen eigentlich immer auch an eine systemische Sklerose gedacht werden sollte. Viele Patient:Innen mit systemischer Sklerose (SSc) haben einen erhöhten ANA-Titer, wobei verschiedene hoch spezifische Antikörper bekannt sind (z.B. gegen Centromer bei der kutan limitierten Form, gegen RNA-Polymerase III und Scl-70 bei der diffusen Form).
Idiopathische inflammatorische Myopathien
Diese sehr seltenen Erkrankungen teilen sich hauptsächlich in die Polymyositis und die Dermatosmyositis. Letztere kann hochcharakteristische Hauterscheinungen, die sogenannten Gottron Papeln aufweisen Klinisch im Vordergrund stehen dabei eine proximale Muskelschwäche, welche v.a. die Kraftausdauer (und insbesondere zu Beginn weniger die Maximalkraft) betrifft und mit Schmerzen einhergehen kann, aber nicht muss. Entsprechend fällt im Labor oft eine erhöhte Kreatininkinase (CK) und ein erhöhtes Myoglobin auf. Der ANA-Titer ist oft, aber nicht immer erhöht und nicht in jedem Fall kann ein spezifischer Antikörper nachgewiesen werden. Je nach klinischem Bild und Antikörper-Profil ist die Prognose zum Teil sehr unterschiedlich (von rasch vital bedrohend bis eher mild). Bei diesen Krankheitsbildern muss immer und insbesondere bei gewisser (Antikörper-) Konstellation an die Möglichkeit eines Tumors gedacht werden und ein Screening hinsichtlich Neoplasien veranlasst werden, da diese hier deutlich gehäuft auftreten.
Mischkollagenose (mixed connective tissue disease, MCTD)
Diese auch als Sharp-Syndrom (Erstbeschreibung durch Sharp 1972) bezeichnete Erkrankung zeigt, wie der Name bereits suggeriert, Charakteristika der anderen Kollagenosen, aber nicht in der Konstellation, als dass ein Overlap-Syndrom diagnostiziert werden kann. Die Eigenständigkeit dieser Krankheit ist teilweise umstritten, da sich im Krankheitsverlauf zuweilen eher das Bild einer SSc oder eines SLE ergeben kann. Im Allgemeinen zeigen die Betroffenen aber eine aggressivere, oft auch destruktive Polyarthritis (erinnernd an eine rheumatoide Arthritis), eine Myositis, diffus geschwollene Finger (sogenannte Puffy Fingers) mit im Verlauf auch möglichem Skleroderm, ein Raynaud Phänomen und im Labor hoch-tritrig positiven ANA (>1:10240) mit spezifisch positiven Antikörpern gegen U1 Ribonucleoprotein (U1 RNP). Wie erwähnt sind auch Nierenbeteiligungen wie bei SLE und Lungenbeteiligungen wie bei SSc (v.a. PAH, aber auch Fibrose) möglich.
Copyright bei Aerzteverlag medinfo AG
Schulthess Klinik
Rheumatologie und Rehabilitation
Lengghalde 2
8008 Zürich
Schulthess Klinik
Rheumatologie und Rehabilitation
Lengghalde 2
8008 Zürich
marco.etter@kws.ch
Die Autoren haben keine Interessenskonflikte im Zusammenghang mit diesem Artikel deklariert.
Auf Anfrage bei den Autoren


der informierte @rzt
- Vol. 13
- Ausgabe 9
- September 2023