

Polyzythämie wird durch einen Anstieg der Hämoglobinwertes (Hb), des Hämatokrits (Hkt) oder Anzahl der roten Blutkörperchen (RBC) über dem Referenzbereich definiert und ist abhängig vom Alter, Geschlecht und Wohnhöhe (üblicherweise ab >1500 m ü.M.). Polyzythämie wurde bei ca. 0.2% aller hospitalisierten Patienten beschrieben (1) und wird in JAK2-mutierte (JAK2-positive) Polyzythämie, welche Polyzythämia vera (PV) genannt wird, sowie JAK2-unmutierte (JAK2-negative) Polyzythämie, welche sekundär, hereditär oder idiopathisch sein kann, eingeteilt.
Polycythaemia is defined by an increase in haemoglobin (Hb), haematocrit (Hct) or red blood cell (RBC) count above the reference range, and is dependent on age, gender and altitude (usually >1500 m above sea level). Polycythaemia has been described in approximately 0.2% of all hospitalised patients (1) and is divided into JAK2-mutated (JAK2-positive) polycythaemia, which is called polycythaemia vera (PV), and JAK2-unmutated (JAK2-negative) polycythaemia, which can be secondary, hereditary or idiopathic.
Key Words: Polycythaemia, haemoglobin, haematocrit, red blood cell¸ JAK2
JAK2-mutierte (JAK2-positive) Polyzythämie: Polyzythämia Vera (PV)
Die PV ist eine hämatologische, klonale Erkrankung, welche zu der Gruppe der myeloproliferativen Neoplasien (MPN) gehört. Die PV-Patienten haben ein deutlich erhöhtes Risiko für venöse und arterielle thromboembolische (auch lebensbedrohliche) Ereignisse. Zudem besteht bei ihnen ein Risiko für die Entwicklung einer Myelofibrose, einer Form der MPN mit einer ungünstigeren Prognose. In seltenen Fällen kann die Erkrankung auch in eine akute myeloische Leukämie (AML) fortschreiten. Gemäss der Klassifikation der Tumorerkrankungen der Weltgesundheitsorganisation (WHO) sowie dem Internationalen Consensus der Klassifikation (ICC) der MPN aus dem Jahr 2022 gibt es klare diagnostische Kriterien für die PV (Tab. 1).

Die JAK2-Mutationen können primär aus dem peripheren Blut getestet werden. Ein erniedrigter Epo-Spiegel ist für PV typisch.
Eine Knochenmarkpunktion (KMP) ist hilfreich, um den Typ der MPN zu charakterisieren und das Vorliegen einer Fibrose, die prognostische Relevanz hat, zu prüfen. Mittels KMP werden Knochenmarkaspirat und Biopsie gewonnen. Wichtige Aspekte bei der Beurteilung sind die gesteigerte Zellularität, eine trilinäre Myeloproliferation, die Morphologie der Megakaryopoese, das Vorhandensein einer Fibrose, sowie eventuelle Vermehrung der Blasten. Die Flowzytometrie ist nicht zwingend nötig, könnte aber dazu beitragen, eine Zunahme der Blasten auszuschliessen. Die konventionelle Zytogenetik ist bei der Diagnosestellung empfohlen, das Material kann jedoch eventuell für spätere Testung asserviert werden. Aufgrund des im Allgemeinen indolenten Verlaufs der PV gehört eine erweiterte molekulare Untersuchung derzeit nicht zur Routine einer Abklärung. Dies ist jedoch in den Fällen mit grenzwertigem Erscheinungsbild, bei denen die Unterscheidung zu anderen MPN schwierig ist, oder bei denen der Verdacht auf ein Fortschreiten der Krankheit besteht, angezeigt. Die erweiterte Charakterisierung erfolgt mittels Next Generation Sequencing (NGS). Es wird ein der Fragestellung entsprechendes Panel eingesetzt. Diese Untersuchung ist jedoch in der Regel mit höheren Kosten verbunden und bedarf einer Zustimmung der Krankenkasse mit einer Kostengutsprache. Der Nachweis zusätzlicher Mutationen kann prognostisch wichtig sein.
Ein Thrombophilie-Screening bei stattgehabter Thrombose kann erwogen werden. Die Testung für Cholesterin (LDL und HDL), Triglyceride, Glucose sowie HbA1c, zur Einschätzung nicht-MPN-bedingter kardiovaskulärer Risikofaktoren sollte erfolgen. Die Optimierung dieser Risikofaktoren ist strikt empfohlen.
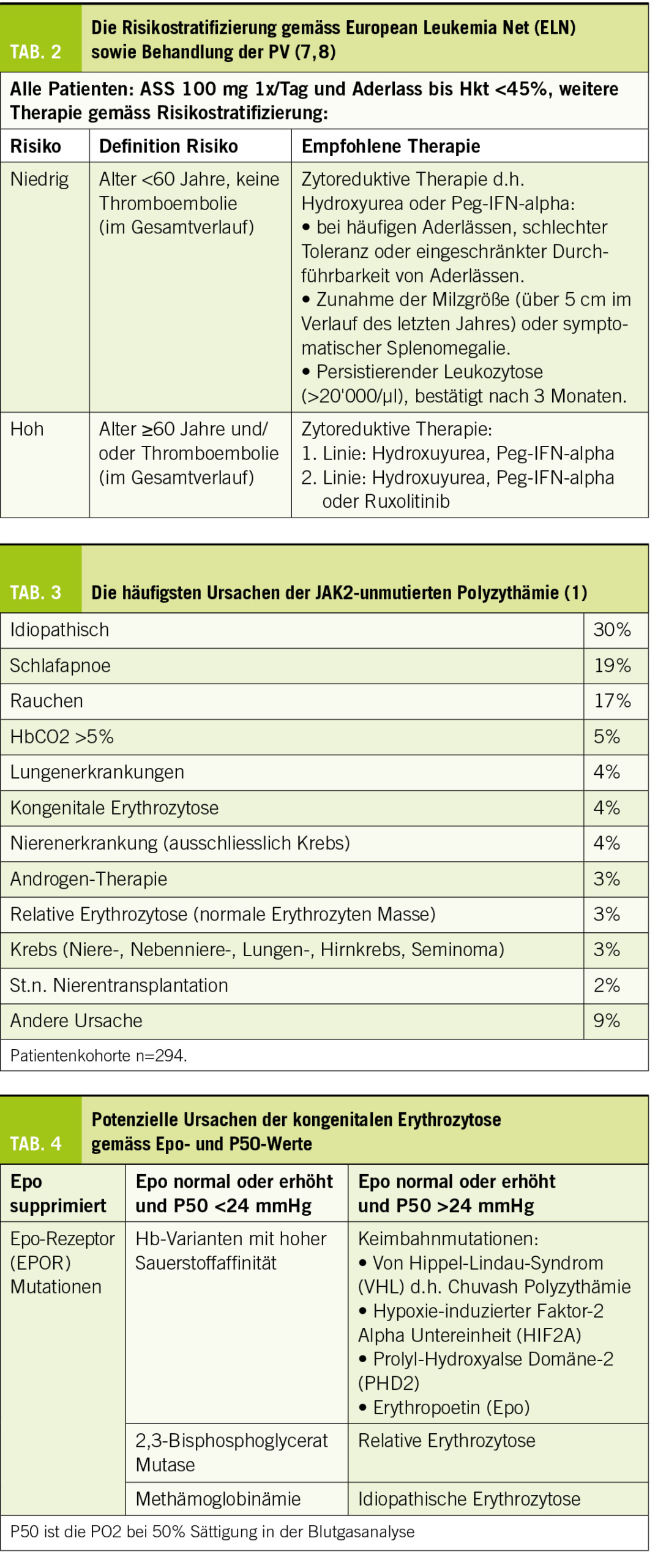
Die Diagnose einer PV hat erhebliche therapeutische Konsequenzen, da aufgrund des erhöhten thromboembolischen Risikos der Hkt <45% gehalten werden soll. Die Behandlung hängt vom Alter und der Vorgeschichte stattgehabter Thrombosen ab, die das Risiko für weitere thromboembolische Komplikationen und die richtige Behandlung der Krankheit bestimmen. Die Behandlung umfasst Aderlässe, Thrombozytenaggregationshemmer d.h. Acetylsalicylsäure (ASS) und zytoreduktive Therapie mit Hydroxyurea oder pegyliertem Interferon alpha (Peg-IFN-alpha) (Tab. 2).
Die Hauptziele der Behandlung sind:
1. Reduktion des thromboembolischen Risikos
2. Kontrolle der klinischen Symptome
3. Vermeidung der späten Komplikationen wie
Myelofibrose oder AML
JAK2-unmutierte (JAK2-negative) Polyzythämie: sekundär, hereditär, idiopathisch
Mit der Entdeckung von JAK2-Treibermutationen im Jahr 2005 und breit verfügbaren Methoden zur Testung stellt die PV meist keine diagnostische Schwierigkeit mehr dar. Eine diagnostische Herausforderung ist dagegen die JAK2-unmutierte d.h. JAK-2 negative Polyzythämie, welche eine heterogene Gruppe von Entitäten umfasst.
In der Universitätsklinik der Hämatologie des Inselspitals Bern wurde eine Studie durchgeführt, in welcher die Epidemiologie, Ursachen und die Komplikationen der JAK2-unmutierten Polyzythämie analysiert wurden (1). In dieser Studie wurden alle Patienten, d.h. ca. 730’000, welche am Inselspital über fast 11 Jahre (Oktober 2008 – Juli 2019) in Behandlung waren, untersucht, und davon ca. 1’400 Patienten, welche die Hb/Hkt WHO Kriterien für PV erfüllt haben, identifiziert. Hiervon wurden 95 Fälle der JAK2-mutierten Polyzythämie d.h. PV, und 294 Fälle mit JAK2-unmutierter Polyzythämie festgestellt. Die Resultate der Studie werden in diesem Artikel diskutiert.
Sekundäre und idiopathische, JAK2-unmutierte Polyzythämie
Prinzipiell treten die erworbenen Formen der JAK2-unmutierten Polyzythämie am häufigsten im klinischen Alltag auf. Sie entstehen als Folge von übermässiger Epo-Produktion, entweder als physiologischer Ausgleich für unzureichende Sauerstoffversorgung des peripheren Gewebes (Lunge), Schlafapnoe, Rechts-Links-Herz-Shunts, Rauchen, inklusive Shisha (2), Höhenlage, Nierenarterienstenose und polyzystische Nierenerkrankung, oder als autonome Produktion von Epo durch Tumore oder die Einnahme von Anabolika (3).
Unsere Studie zeigt, dass die Männer der Kohorte der JAK2-unmutierten Polyzythämie stark überrepräsentiert waren (n=242, 82%).
Trotz der Verfügbarkeit molekularer Untersuchungen und anderer diagnostischer Methoden sind idiopathische Formen nach wie vor die häufigste Ursache in dieser Krankheitsgruppe, sodass bei 30 % dieser Patienten keine Ursache festgestellt wurde, gefolgt von Schlafapnoe und Rauchen (Tab. 3).
Bei jungen Patienten (< 30 J., n=56), liess sich eine Ursache der Polyzythämie nur bei ca. der Hälfte eruieren. Bei diesen Patienten blieb die Polyzythämie, trotz extensiver Diagnostik, unklar. Darüber hinaus sind bei jungen Patienten die Schlafapnoe (n=6, 11% aller jungen Patienten) und Rauchen (n=6, 11% aller jungen Patienten) die am häufigsten diagnostizierbaren Ursachen für JAK2-unmutierte Polyzythämie. Aus diesem Grund sollte die Schlafapnoe auch bei jungen Patienten aktiv gesucht werden.
Die sekundäre und die idiopathische Polyzythämie benötigen aus hämatologischer Sicht keine Therapie, da diese Patienten prinzipiell kein klar erhöhtes Risiko für Thromboembolien oder Transformation in eine andere Erkrankung haben. Die Ursache sollte therapeutisch angegangen werden.
Kongenitale Erythrozytose
Angeborene sekundäre Erythrozytosen sind selten und werden meist durch Keimbahnmutationen verursacht, einschliesslich Mutationen in Genen, die am Sauerstoff-Sensorweg und Hb-Bildung beteiligt sind, unter anderem abnormale Sauerstoffaffinität (4). Es sind mehrere Mutationen bekannt, welche eine kongenitale Erythrozytose verursachen können (Tab. 4).
Parameter, die für die Kategorisierung dieser Art von Polyzythämie wegweisend sind, umfassen den Epo-Spiegel im Serum und den P50-Wert. Ein niedriger P50-Wert (PO2 bei 50% Sättigung) in der Blutgasanalyse spiegelt eine hohe Hb-Sauerstoffaffinität wider (5). In unserer Erfahrung ist jedoch die Testung für Epo und P50 in der arteriellen (oder venösen) Blutgasanalyse, selten hilfreich in der Feststellung der Ursache der kongenitalen Erythrozytose, da die bestimmten Mutationen mit verschiedenen Mustern der Epo und P50 Werte verbunden sind (1).
Um die Mutation, welche für die kongenitale Erythrozytose verantwortlich ist, zu untersuchen, muss eine molekulare Testung durchgeführt werden. Diese kann vom peripherenBlut erfolgen und mit NGS durchgeführt werden. NGS ist eine molekulardiagnostische Methode, die erlaubt, mehrere Gene gleichzeitig zu untersuchen und auch vor einer KMP durchgeführt werden kann.
Am Inselspital wurde bereits im Jahr 2018, ein 13-Gen-NGS-Panel für die kongenitale Erythrozytose entwickelt (6). Dieses Panel wurde über die letzten Jahre um weitere Gene erweitert und aktuell werden 24 Gene untersucht (Tab. 5).
Es werden nur Genvarianten untersucht, die gemäß den internationalen Richtlinien als pathogen, wahrscheinlich pathogen oder als Varianten unbekannter Signifikanz (VUS) gelten.
Diese Untersuchung ist vor allem bei jungen Patienten oder Patienten mit positiver Familienanamnese indiziert und benötigt eine Kostengutsprache der Krankenkasse.
Die kongenitalen Erythrozytosen benötigen aus hämatologischer Sicht ebenfalls meistens keine spezifische Therapie und sind nicht mit erhöhtem Risiko für Thrombosen oder Transformation in eine andere Erkrankung verbunden. Die Ausnahme ist die Chuvah Erythrozytose (VHL-mutiert), da diese für eine Thrombosetendenz bekannt ist. Aktuell gibt es jedoch keine klare Therapieempfehlung. Mit der Zunahme der Testung für die kongenitalen Erythrozytosen sowie einer besseren Datenlage, können hoffentlich zukünftig die klaren Managementstrategien etabliert werden.

Diagnostisches Vorgehen bei Patienten mit Erythrozytose
Die Schrittempfehlungen für die Abklärung bei Patienten mit Erythrozytose/ Polyzythämie, die negativ für JAK2-Mutationen in Exon 14 und 12 ist, sind:
1. Genaue Anamnese und Vorgeschichte: Vorerkrankungen? Allgemeine Symptome? Rauchen? Medikamente (Androgen-, Epo-Einnahme)? Gezielte Anamnese bzg. Tumorerkrankungen? Nierentransplantation? Überprüfung früherer Blutbild-Werte: Hatte der Patient jemals normale Hb-/Hkt-Werte?
2. Zuweisung in die Pneumologie zur Abklärung der Schlafapnoe oder anderen Lungenerkrankungen (auch bei beschwerdefreien Patienten). Röntgenuntersuchung des Thorax, Lungenfunktionsprüfung erwägen. Messung von COHb.
3. Screening der kardialen Erkrankungen mit Echokardiogramm sowie Elektrokardiogramm (EKG), wenn noch nicht erfolgt. Zuweisung Kardiologie erwägen.
4. Ultraschall Abdomen (Frage nach Hepatosplenomegalie, Lymphadenopathie, Tumormasse, Nierenarterienstenose, Nierenzysten) und/oder gezielte Bildgebung zur Tumorabklärung je nach Symptomatik.
5. Altersentsprechendes Tumorscreening gemäss Standard.
In fast einem Drittel der Patienten bleibt jedoch die Ursache einer JAK2-unmutierten Polyzythämie trotz erweiterter Suche nach sekundären oder kongenitalen Ursachen unklar und wird als idiopathisch klassifiziert.
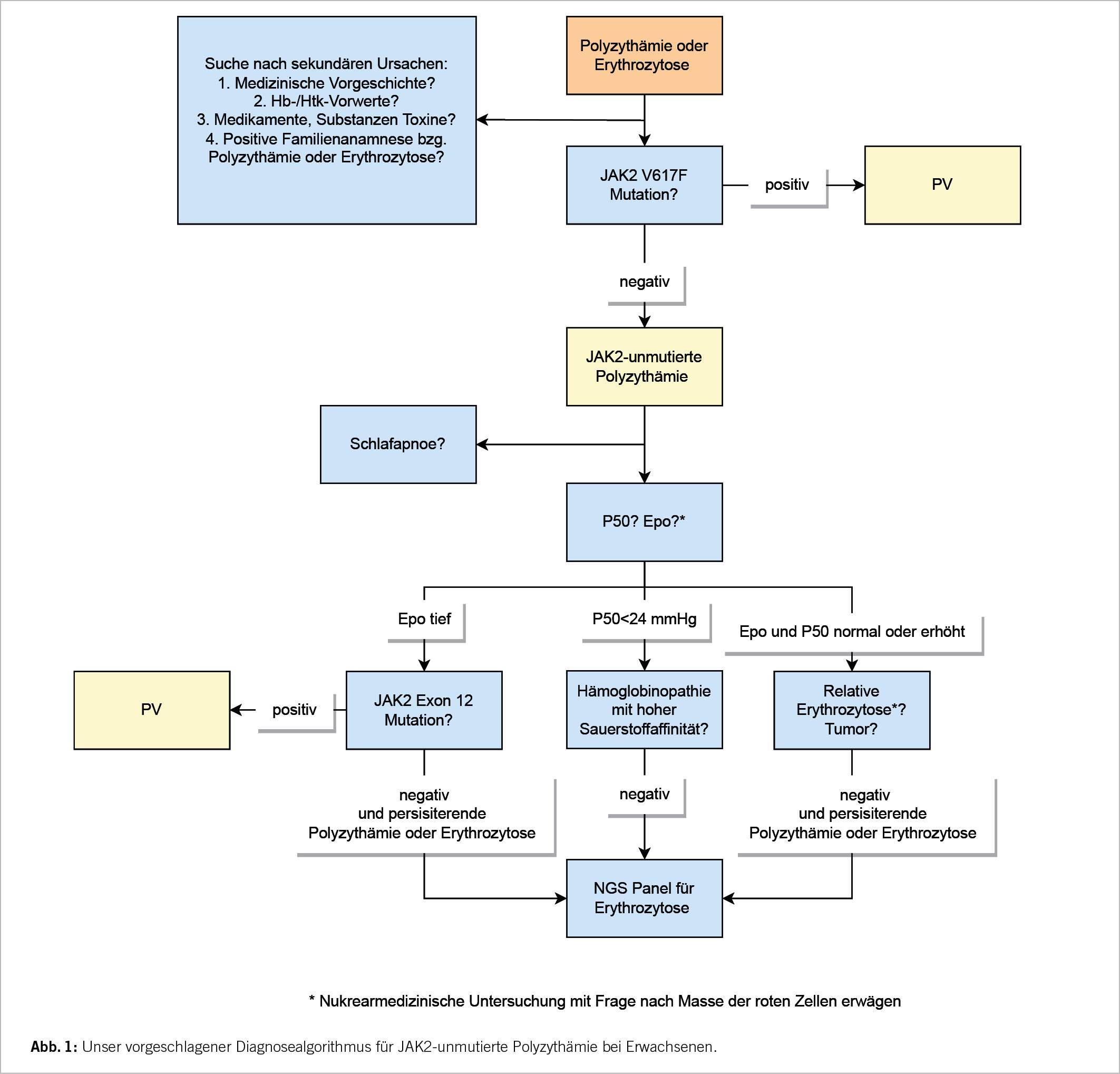
Unser vorgeschlagener Diagnosealgorithmus für JAK2-unmutierte Polyzythämie bei Erwachsenen ist in Abbildung 1 dargestellt.
Zusammenfassend kann gesagt werden, dass die Polyzythämie und Erythrozytose wichtige Laborbefunde sind, welche eine weitere Abklärung triggern sollten.
Danksagung: Herzlichen Dank an die Jacques und Gloria Gossweiler Stiftung (www.jggf.ch), welche die Forschung über JAK2-unmutierte Polyzythämie im Rahmen des Fellowship-Preises 2020 für K. A. Jalowiec unterstützt hat.
Copyright bei Aerzteverlag medinfo AG
Dr. med. Katarzyna Aleksandra Jalowiec, MSc
Dr. sc. nat. Naomi Porret
Prof. Dr. med. Sara Christina Meyer, PhD
Prof. Dr. med. Alicia Rovó
Universitätsklinik für Hämatologie und
Hämatologisches Zentrallabor
Inselspital, Universitätsspital Bern, Freiburgstrasse, 3010 Bern
Universitätsklinik für Hämatologie und
Hämatologisches Zentrallabor
Inselspital, Universitätsspital Bern
Freiburgstrasse
3010 Bern
Universitätsklinik für Hämatologie und
Hämatologisches Zentrallabor
Inselspital, Universitätsspital Bern
Freiburgstrasse
3010 Bern
Universitätsklinik für Hämatologie und
Hämatologisches Zentrallabor
Inselspital, Universitätsspital Bern
Freiburgstrasse
3010 Bern
Die Autorinnen haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Jalowiec, K.A. et al. (2022) JAK2 Unmutated Polycythaemia—Real-World Data of 10 Years from a Tertiary Reference Hospital. Journal of Clinical Medicine [online]. 11 (12), p. 3393.
2. Agbariah, N. and Rovó, A. (2022) Breaking Stereotypes: Polycythemia Secondary to Shisha Smoking in a Middle-Age Swiss Woman. Acta Haematologica [online]. 145 (6), pp. 650–654.
3. Gangat, N., Szuber, N., Pardanani, A. and Tefferi, A. (2021) JAK2 unmutated erythrocytosis: current diagnostic approach and therapeutic views. Leukemia [online]. 35 (8), pp. 2166–2181.
4. McMullin, M.F. (2016) Investigation and Management of Erythrocytosis. Current Hematologic Malignancy Reports [online]. 11 (5), pp. 342–347.
5. Perroud, C., Porret, N. and Rovó, A. (2023) ‘The Long Journey of Unexplained Erythrocytosis’: Erythrocytosis due to High-Oxygen Affinity Hemoglobinopathy – Hemoglobin Variant Little Rock (HBB: c.432C>A) – A Report of a Swiss Family and Review of the Literature. Acta Haematologica [online]. 146 (4), pp. 326–330.
6. Jalowiec, K.A., Vrotniakaite-Bajerciene, K., Capraru, A., Wojtovicova, T., Joncourt, R., Rovó, A. and Porret, N.A. (2021) NGS Evaluation of a Bernese Cohort of Unexplained Erythrocytosis Patients. Genes [online]. 12 (12), p. 1951.
7. Marchetti, M. et al. (2022) Appropriate management of polycythaemia vera with cytoreductive drug therapy: European LeukemiaNet 2021 recommendations. The Lancet Haematology [online]. 9 (4), pp. e301–e311.
8. Barbui, T. et al. (2018) Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia [online]. 32 (5), pp. 1057–1069.



der informierte @rzt
- Vol. 14
- Ausgabe 3
- März 2024