

Polypharmazie ist der wichtigste Risikofaktor für das Auftreten von Arzneimittelinteraktionen. Patienten mit Herzinsuffizienz oder Hypertonie haben oft eine Polypharmazie, wenn sie gemäss Leitlinien behandelt werden. Entsprechend hoch ist die Prävalenz von Arzneimittelinteraktionen bei diesen Patienten. Arzneimittelinteraktionen können die Pharmakokinetik von Arzneistoffen verändern, was meistens eine Folge der Hemmung oder Induktion von Enzymen zum Arzneistoffmetabolismus oder von Proteinen zum Arzneistofftransport ist. Daneben gibt es pharmakodynamische Interaktionen, wenn Arzneistoffe mit ähnlicher Wirkung aber unterschiedlichem Mechanismus kombiniert werden. Im Artikel gehe ich auf die wichtigsten Interaktionen von Arzneistoffklassen/Arzneistoffen ein, welche bei der Therapie der Herzinsuffizienz oder Hypertonie eine Rolle spielen.
Polypharmacy is the most important risk factor for the occurrence of drug interactions. Patients with heart failure or hypertension often have polypharmacy if they are treated according to guidelines. The prevalence of drug interactions is correspondingly high in these patients. Drug interactions can alter the pharmacokinetics of drugs, which is usually a result of the inhibition or induction of enzymes for drug metabolism or proteins for drug transport. There are also pharmacodynamic interactions when drugs with similar effects but different mechanisms are combined. In this article, I will discuss the most important interactions of drug classes/drugs that play a role in the treatment of heart failure or hypertension.
Key words: Polypharmacy, drug interactions, pharmacokinetics of drugs, heart failure, hypertension
Arzneistoffinteraktionen sind bei internistischen Patienten häufig anzutreffen. In einer von uns durchgeführten Studie hatten bei Spitaleintritt auf eine internistische Station ca. 30% der Patienten eine Arzneistoffinteraktion, die zu einer klinisch relevanten unerwünschten Wirkung hätte führen können (Vonbach et al., 2008). Bei Patienten mit dekompensierter Herzinsuffizienz fanden wir bei Entlassung aus dem Spital bei 89% eine Arzneimittelinteraktion, die meisten davon wurden hoch- oder mittelgradig relevant beurteilt (Straubhaar et al., 2006). Arzneistoffinteraktionen sind ein wichtiger Risikofaktor für das Entstehen von unerwünschten Wirkungen, aber nur bei einer Minderheit der Interaktionen treten unerwünschte Wirkungen auf (Egger et al., 2010). Trotzdem ist es wichtig, Interaktionen zu erkennen, das Risiko für das Auftreten sowie die möglichen Folgen einer unerwünschten Wirkung mit dem erwarteten Therapieerfolg zu vergleichen und zu entscheiden, ob die Interaktion toleriert oder aufgehoben werden soll. Der wichtigste Risikofaktor für das Auftreten von Interaktionen ist die Polypharmazie (Rätz Bravo et al., 2005), welche gerade bei Patienten mit Herzinsuffizienz oder Hypertonie oft vorkommt (Straubhaar et al., 2006). Allerdings wird in diesen Patientengruppen die Pharmakotherapie meist gemäss Richtlinien durchgeführt und ist deshalb standardisiert, was das Risiko für relevante Interaktionen senkt. Ein weiterer wichtiger Risikofaktor für das Auftreten von relevanten Interaktionen ist die Behandlung desselben Patienten durch verschiedene Ärzte (Tamblyn et al., 1996), was die Wichtigkeit einer guten Kommunikation zwischen den behandelnden Ärzten und die zentrale Rolle der Hausärzte bei der Vermeidung, Erkennung und dem Umgang mit Arzneistoffinteraktionen unterstreicht.
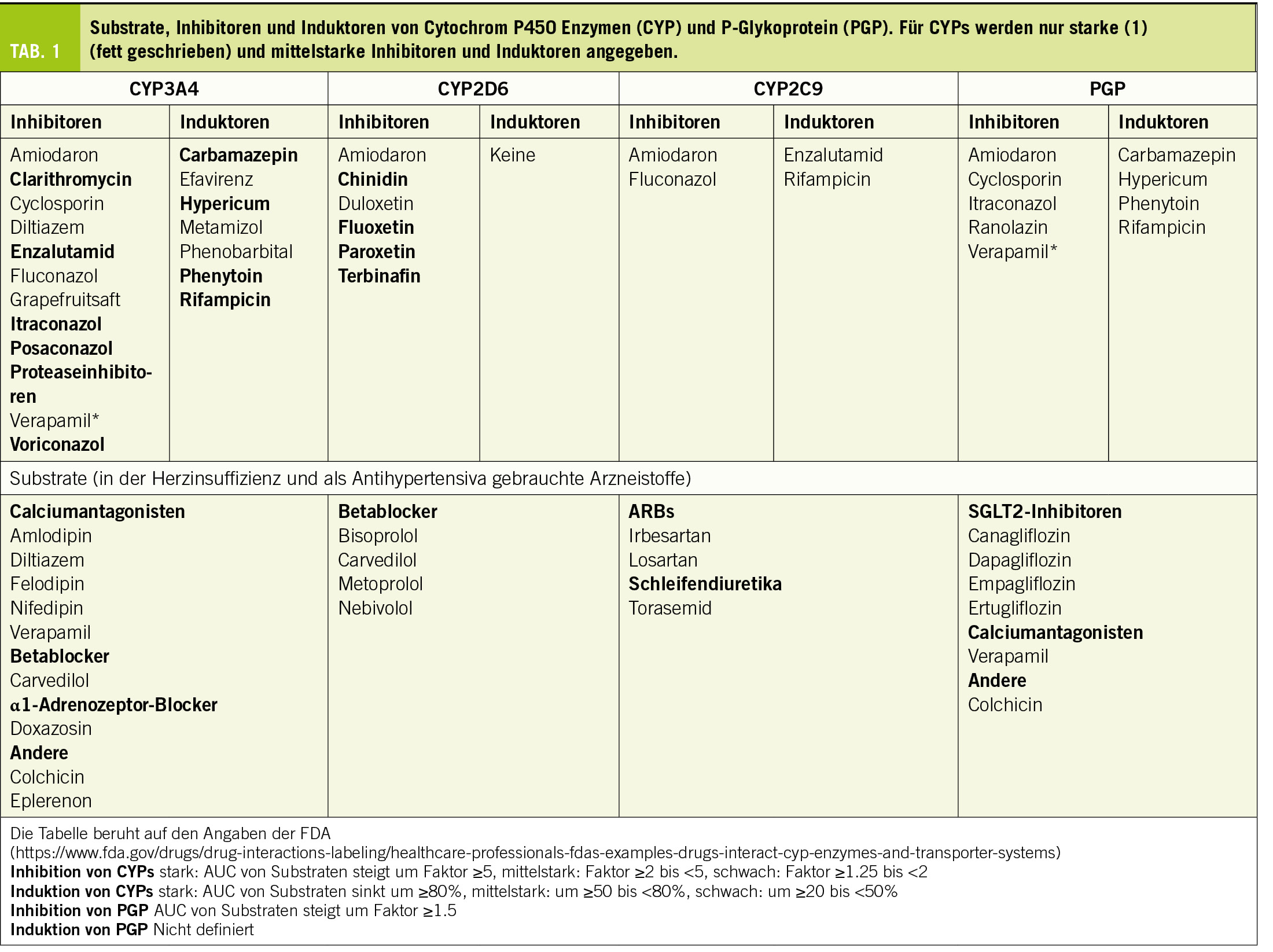
Bezüglich des Mechanismus von Arzneistoffinteraktionen kann grob zwischen pharmakokinetischen und pharmakodynamischen Interaktionen unterschieden werden. Bei den pharmakokinetischen Interaktionen verändert sich die Plasmakonzentration des «Opfers» der Interaktion, währenddem bei pharmakodynamischen Interaktionen eine Kombination von Arzneistoffen mit ähnlichen pharmakodynamischen Eigenschaften (aber unterschiedlichem Wirkmechanismus) vorliegt. Die pharmakokinetischen Interaktionen beruhen meist auf der Hemmung oder Induktion des für den Abbau eines bestimmten Arzneistoffes verantwortlichen Enzyms oder des für den Transport des Arzneistoffes verantwortlichen Proteins. Die wichtigsten für den Arzneistoffmetabolismus verantwortlichen Enzyme, die Cytochrom P450 Enzyme (CYPs) und der wichtigste Arzneistofftransporter, das P-Glykoprotein (PGP) sind in der Tabelle 1 beschrieben. Im Gegensatz zu den pharmakokinetischen sind die pharmakodynamischen Interaktionen in der Kardiologie oft erwünscht, so z.B. bei der Kombination unterschiedlicher Antihypertensiva oder von Statinen mit Ezetimib zur Senkung des LDL-Cholesterins. Ob eine Interaktion klinisch relevant ist oder nicht hängt im Wesentlichen von der unerwünschten Wirkung ab, welche als Folge einer bestimmten Interaktion auftreten kann.
In der Folge gehe ich auf klinisch relevante Interaktionen bei Patienten mit Herzinsuffizienz und bei Patienten mit Hypertonie ein. Zusätzlich beschreibe ich auch die relevanten Interaktionen von Colchicin, welches zunehmend häufig bei Patienten mit Peri- und/oder Myokarditis eingesetzt wird. Die Interaktionen von Arzneistoffen in anderen Gebieten der Kardiologie (Therapie und Prophylaxe der koronaren Herzkrankheit und Embolieprophylaxe bei Vorhofflimmern) werden in einem Folgeartikel besprochen werden.
Interaktionen mit Arzneistoffen zur Therapie der Herzinsuffizienz
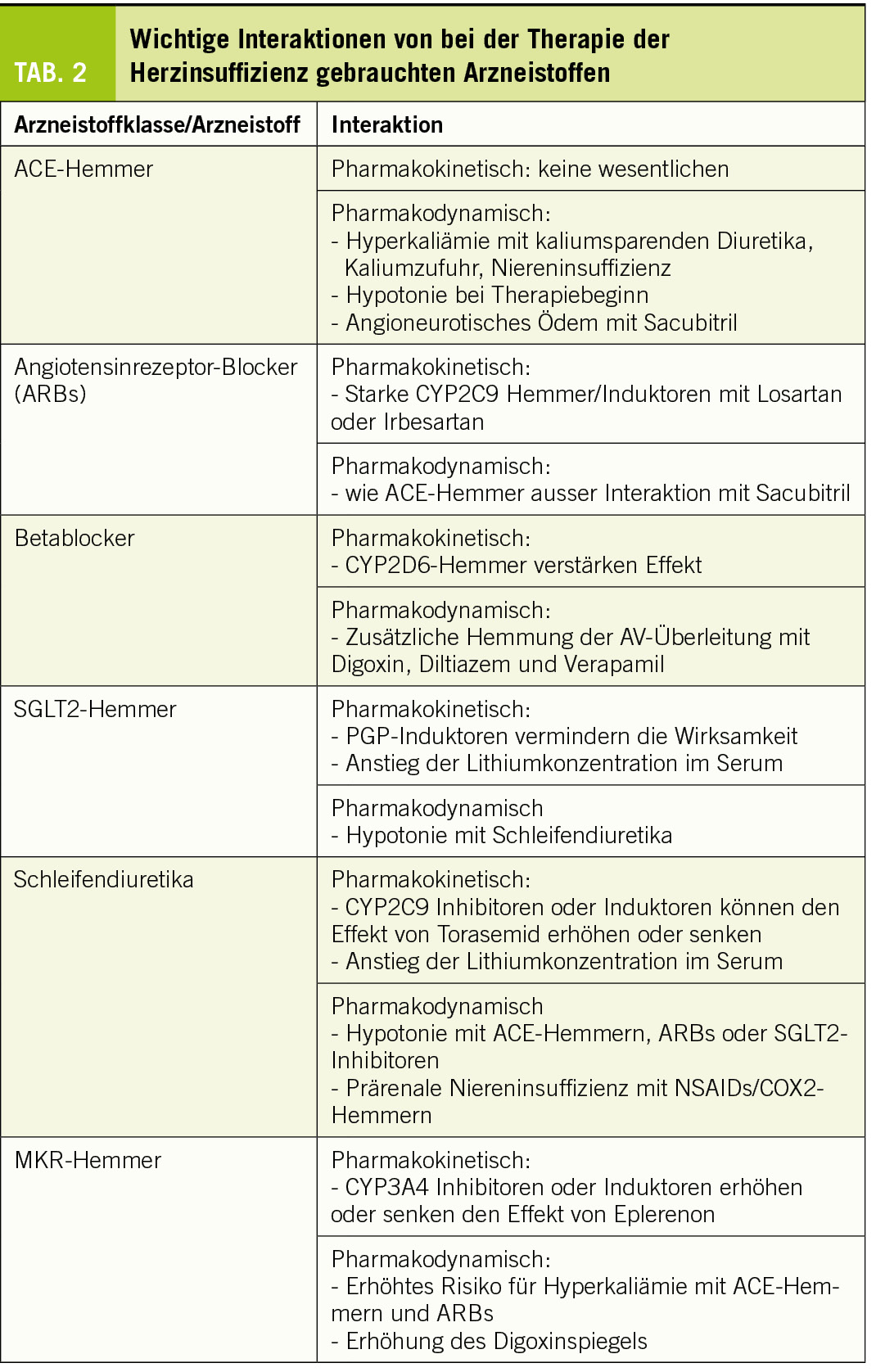
Wichtige Arzneistoffgruppen, welche bei der Therapie von Patienten mit Herzinsuffizienz gebraucht werden, sind ACE-Hemmer, Angiotensinrezeptorblocker (ARBs), Betablocker, SGLT2-Hemmer, Mineralkortikoidrezeptor-Hemmer (MKR-Hemmer) und Schleifendiuretika. Die wichtigsten Interaktionen von Arzneistoffen für die Therapie der Herzinsuffizienz sind in Tabelle 2 gelistet.
ACE-Hemmer
ACE-Hemmer werden als Ester (Ester sind Prodrugs, welche durch Esterasen zu den entsprechenden Säuren hydrolysiert werden) oder Säuren verabreicht, welche in der Regel nicht metabolisiert renal und/oder biliär ausgeschieden werden. Pharmakokinetische Interaktionen via CYPs oder PGP sind deshalb nicht zu erwarten. In der Literatur sind Fallberichte bezüglich Hemmung der renalen Ausscheidung von Lithium zu finden. Allerdings lag bei diesen Patienten oft auch eine Niereninsuffizienz (eventuell verschlimmert durch Akkumulation von vorwiegend renal eliminierten ACE-Hemmern wie Lisinopril) oder eine gleichzeitige Verabreichung von Thiaziden vor (Hommers et al., 2019). Unter Kontrolle der Nierenfunktion und Vermeidung von Arzneistoffen, welche die Elimination von Lithium behindern (wie Thiazide) sowie regelmässiger Bestimmung des Lithiumspiegels können ACE-Hemmer aus meiner Sicht gleichzeitig mit Lithium verabreicht werden.
Zu den pharmakodynamischen Interaktionen gehören der mögliche Blutdruckabfall zu Beginn der Therapie mit einem ACE-Hemmer bei Patienten, welche schon mit einem Schleifendiuretikum behandelt werden sowie die Hyperkaliämie bei Kombination mit MKR-Hemmern und/oder Zufuhr von Kalium. Die initiale Hypotonie kann durch vorsichtige Eintitration des ACE-Hemmers vermieden werden. ACE-Hemmer und ARBs gehören zur etablierten Therapie bei Herzinsuffizienz mit verminderter LVEF (HFrEF), ebenso die MKR-Hemmer Spironolacton und Eplerenon. Da sowohl ARBs/ACE-Hemmer und MKR-Hemmer durch Verminderung der Wirkung von Aldosteron zu einer Hyperkaliämie führen können, entsteht bei gleichzeitiger Einnahme eine pharmakodynamische Interaktion. Diese führt nur selten zur Hyperkaliämie, wenn die Patienten engmaschig überwacht werden. Faktoren, welche das Auftreten einer Hyperkaliämie bei solchen Patienten wahrscheinlicher machen, sind die gleichzeitige Zufuhr von Kalium und eine Niereninsuffizienz. Dem Auftreten einer Hyperkaliämie bei mit ACE-Hemmern/ARBs und MKR-Hemmern behandelten Patienten liegt dementsprechend oft eine prärenale Niereninsuffizienz zugrunde, sei es durch ungenügende Flüssigkeitszufuhr, Wasserverlusten durch Erbrechen/Diarrhöe oder durch überdosierte Schleifendiuretika. Die Nierenfunktion muss deshalb bei mit ACE-Hemmern/ARBS und MKR-Hemmern behandelten Patienten gut überwacht werden, insbesondere wenn weitere Risikofaktoren für eine Hyperkaliämie dazukommen.
Die Kombination des Neprilysinhemmers Sacubitril mit ACE-Hemmern muss vermieden werden, da es darunter zu einem Anstieg von Bradykinin und damit zu angioneurotischem Ödem kommen kann. Bei einem Wechsel von einem ACE-Hemmer auf die Kombination ARB/Sacubitrilmuss deshalb ein genügender zeitlicher Abstand (im Kompendium steht 36 Stunden, besser sind 5 Halbwertszeiten des ACE-Hemmers) eingehalten werden.
Betablocker
Bei der Therapie der Herzinsuffizienz gebraucht werden die kardioselektiven Betablocker Bisoprolol, Metoprolol und Nebivolol (Block der β1-Adrenozeptoren) und das nicht-selektive Carvedilol (Block der β1-, β2- und α1-Adrenozeptoren). Die stark lipophilen Betablocker Nebivolol und Carvedilol werden zu fast 100% metabolisiert (CYP2D6 für Nebivolol und CYP3A4, 2D6, 2A1 und 2C9 für Carvedilol). Bisoprolol und Metoprolol sind weniger lipophil und werden zu 50% (Bisoprolol) oder 40% (Metoprolol) unverändert renal ausgeschieden. Der Rest wird via CYP2D6 oxidiert und vorwiegend renal ausgeschieden.
CYP2D6 ist das Hauptsächlichste an der Metabolisierung von Betablockern beteiligte CYP. CYP2D6 kann nicht induziert, sondern nur gehemmt werden (Berger et al., 2016). Die wichtigsten Hemmer von CYP2D6 sind in Tabelle 1. aufgelistet. Relevant könnte die gleichzeitige Verabreichung eines starken CYP2D6-Hemmers vor allem für Nebivolol sein, da Nebivolol zu fast 100% via CYP2D6 metabolisiert wird. Der Effekt von Nebivolol wird in diesem Fall zunehmen, was klinisch an der daraus resultierenden Bradykardie und allenfalls Hypotonie gut erkennbar ist. Für die Wirkung von Metoprolol und Bisoprolol wird eine Hemmung von CYP2D6 weniger relevant sein, da ein beträchtlicher Anteil unverändert renal eliminiert wird. Beim Abbau von Carvedilol sind verschiedene CYPs beteiligt. Entsprechend steigt die Exposition nach Verabreichung des CYP2D6 Blockers Paroxetin um ca. 70% und nach Amiodaron (mittelstarker Blocker von CYP3A4, 2D6 und 2C9) um einen Faktor von ca. 2, was klinisch relevant ist.
Bei den Betablockern sind nebst den pharmakokinetischen auch die pharmakodynamischen Interaktionen erwähnenswert. Nichtselektive Betablocker hemmen durch Block der β2-Adrenozeptoren die Glykogenolyse (Abbau von Glykogen stimuliert via Aktivierung von β2-Adrenozeptoren durch Adrenalin) und damit eine Gegenregulation bei Hypoglykämie. Zudem hemmen alle Betablocker das Auftreten einer Tachykardie bei Hypoglykämie. Wenn Betablocker bei Diabetikern angewendet werden, sollten also kardioselektive gewählt werden, um nicht das Risiko für Hypoglykämien zu erhöhen. Zudem sollten Patienten darauf hingewiesen werden, dass die Symptome einer Hypoglykämie verschleiert sein können. Da Betablocker negativ inotrop wirken und die AV-Überleitung hemmen, verstärken sie diese Effekte bei den gleichzeitig verabreichten Calciumantagonisten Verapamil und Diltiazem. Der negative Effekt auf die AV-Überleitung muss auch bei einer Kombination mit Digoxin oder Amiodaron beachtet werden. Wie oben beschrieben, führt Amiodaron via CYP-Hemmung zusätzlich noch zu einer höheren Exposition von v.a. Carvedilol. Die Effekte auf die AV-Überleitung können natürlich erwünscht sein, wichtig ist das Erkennen der Interaktion und die Kontrolle der so behandelten Patienten.
SGLT2-Hemmer
In der Schweiz sind gegenwärtig die 4 SGLT2-Inhibitoren Canagliflozin, Dapagliflozin, Empagliflozin und Ertugliflozin im Handel. SGLT2-Inhibitoren haben eine gute Bioverfügbarkeit (≥65%) eine hohe Proteinbindung (≥86%) und werden vorwiegend glukuronidiert, aber nicht via CYPs abgebaut. Alle sind PGP-Substrate, weshalb bei gleichzeitiger Anwendung von PGP-Induktoren (siehe Tabelle 1), die Exposition (AUC) der Gliflozine sinkt (um 40-50% für Canagliflozin und Ertugliflozin). Die Hemmung von PGP spielt demgegenüber fast keine Rolle, weil die Bioverfügbarkeit schon fast 100% beträgt und nicht nennenswert gesteigert werden kann.
Alle SGLT2-Hemmer senken die Lithiumkonzentration im Serum, was klinisch bedeutsam sein kann. Eine erwartete pharmakodynamische Interaktion ist ein erhöhtes Risiko für Dehydration bei mit Schleifendiuretika behandelten Patienten.
Schleifendiuretika
Die Schleifendiuretika Furosemid und Torasemid haben eine gute orale Bioverfügbarkeit (≥65%) und werden vorwiegend renal mittels Filtration und Sekretion ausgeschieden. Furosemid wird zu einem geringen Anteil glukuronidiert (ca. 15%), währenddem Torasemid zu ca. 75% oxidativ (CYP2C9 und 2C8) abgebaut wird. Enzyminduktoren wie z.B. Rifampicin könnten deshalb den diuretischen Effekt von Torasemid vermindern. Beide Schleifendiuretika erhöhen die Lithiumkonzentration im Serum, was beachtet werden muss.
Bedeutsam sind auch die pharmakodynamischen Interaktionen wie Verstärkung des Effekts von Digoxin bei Hypokaliämie, mögliche Hypotonie in Kombination mit ACE-Hemmern oder ARBs bei Therapiebeginn und v.a. das Auftreten einer Niereninsuffizienz in Kombination mit NSAR inklusive COX2-Inhibitoren. Bezüglich Niereninsuffizienz sind v.a. Patienten gefährdet, welche dehydriert und zum Offenhalten der glomerulären Durchblutung auf renal produzierte Prostaglandine angewiesen sind.
Mineralkortikoidrezeptor-Hemmer (MKR-Hemmer)
Dazu gehören Spironolacton und Eplerenon. Spironolacton hat eine fast 100%-ige orale Bioverfügbarkeit und wird via Hydrolyse des Lactonrings zu Canrenon umgewandelt, welches das wirksame Prinzip darstellt. Canrenon wird vorwiegend unverändert renal via Filtration und Sekretion eliminiert. Die Bioverfügbarkeit von Eplerenon beträgt ca. 70%. Im Gegensatz zu Spironolacton ist Eplerenon kein Prodrug; es wird zu ca. 70% renal eliminiert, aber nur etwa 5% als unveränderte Substanz. Der Metabolismus geschieht v.a. via Oxidation durch CYP3A4.
Die Interaktion von Spironolacton und Eplerenon mit ACE-Inhibitoren und ARBs, welche eine Hyperkaliämie begünstigt, ist schon dort beschrieben. Zudem hemmen Spironolacton (Waldorff et al., 1983) und Eplerenon die tubuläre Sekretion von Digoxin, der Digoxinspiegel steigt um ca. 20%. Interaktionen mit CYP-Hemmern und Induktoren spielen nur für Eplerenon eine Rolle. Bei gleichzeitiger Anwendung von starken CYP3A4-Inhibitoren erhöht sich die Exposition von Eplerenon (AUC) um mehr als den Faktor 2, weshalb diese laut Kompendium kontraindiziert sind. Entsprechend fällt die AUC von Eplerenon bei gleichzeitiger Einnahme von CYP3A4 Induktoren. Bei einer Langzeittherapie mit CYP3A4 Induktoren empfiehlt sich deshalb ein Wechsel von Eplerenon auf Spironolacton.
Interaktionen mit Arzneistoffen zur Therapie der Hypertonie
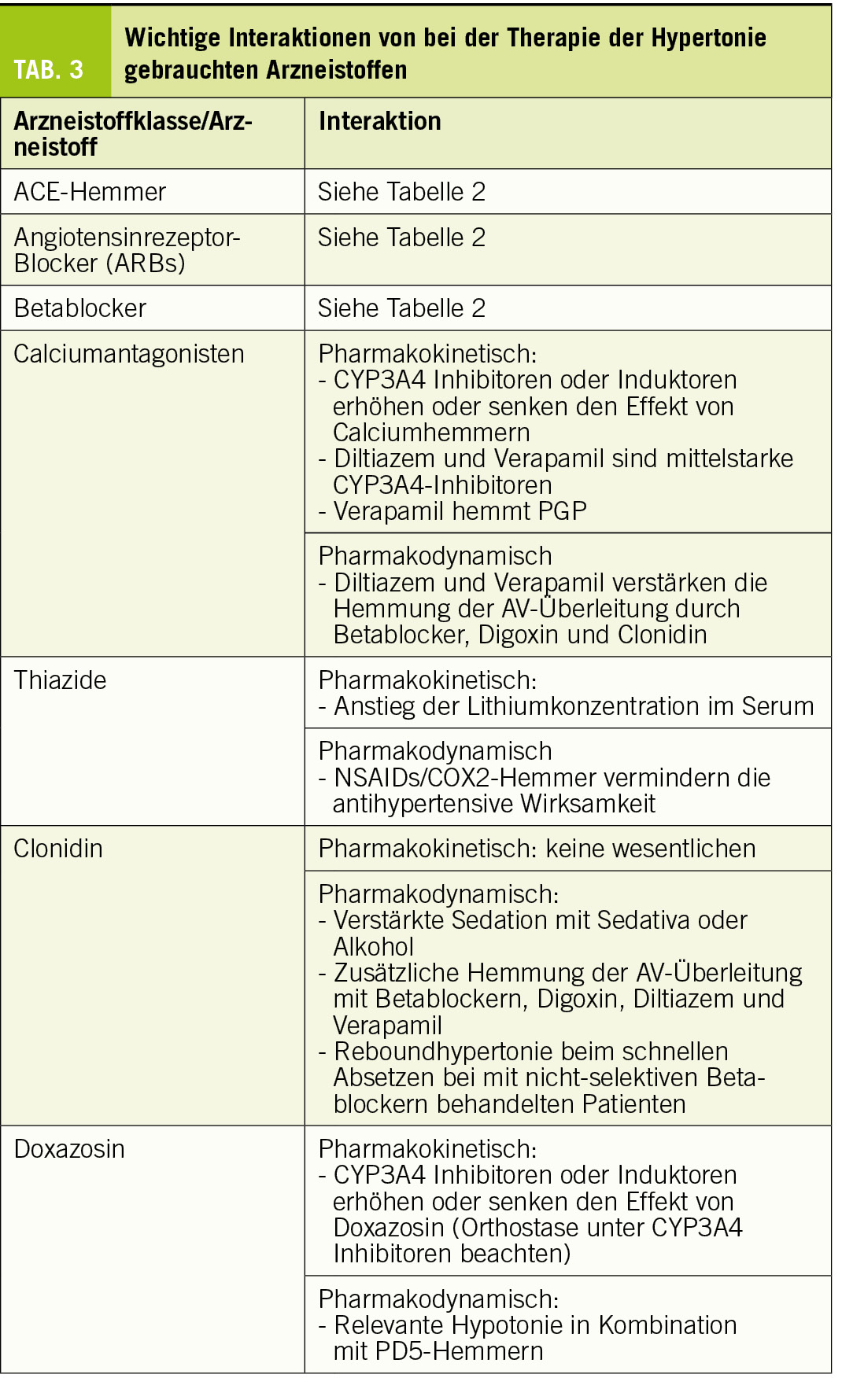
Wichtige Arzneistoffgruppen, welche bei der Therapie von Patienten mit Hypertonie gebraucht werden, sind ACE-Hemmer, Angiotensinrezeptorblocker (ARBs), Calciumantagonisten, Thiazide, Betablocker, Mineralkortikoidrezeptor-Hemmer (MKR-Hemmer), α2-Adrenozeptoren-Agonisten und α1-Adrenozeptoren-Blocker. ACE-Hemmer, ARBs, Betablocker und MKR-Hemmer sind bereits bei der Therapie der Herzinsuffizienz besprochen worden. Die wichtigsten Interaktionen von Arzneistoffen für die Therapie der Hypertonie sind in Tabelle 3 gelistet.
Calciumantagonisten
Alle Calciumantagonisten sind lipophile Substanzen, welche fast vollständig metabolisiert werden. CYP3A4 ist das wichtigste Enzym, welches am Metabolismus von Calciumantagonisten beteiligt ist, was die meisten pharmakokinetischen Interaktionen dieser Arzneistoffgruppe erklärt. Aufgrund der Struktur können die Dihydropyridine (Amlodipin, Felodipin, Isradipin, Lercanidipin und Nifedipin) von Verapamil und Diltiazem unterschieden werden. Mit Ausnahme von Amlodipin und Diltiazem haben die Calciumantagonisten eine Bioverfügbarkeit von <50%, bedingt durch einen relevanten first-pass Effekt. Der Abbau erfolgt dabei schon im Darm (die Dünndarmepithelien haben eine hohe Expression von CYP3A4), was durch einen relevanten Anstieg der Exposition nach Einnahme von Grapefruitsaft gezeigt werden kann (Dresser et al., 2000) (Grapefruitsaft hemmt CYP3A4 nur im Darm, aber nicht in der Leber (Kupferschmidt et al., 1995)). CYP3A4-Hemmer steigern die Exposition und Wirkung aller Calciumantagonisten, wobei der Effekt auf Diltiazem und Amlodipin weniger gross ist als derjenige auf die übrigen Calciumantagonisten. Demgegenüber senken CYP3A4 Induktoren Exposition und Wirkung aller Calciumantagonisten. Da der Blutdruck gut überwacht werden kann, spielen diese Interaktionen klinisch eine eher untergeordnete Rolle.
Diltiazem und Verapamil (aber nicht die Dihydropyridine) sind gleichzeitig auch mittelstarke Hemmer von CYP3A4, was klinisch relevant sein kann und beachtet werden sollte. Verapamil hemmt zudem PGP, was die Exposition von PGP-Substraten wie z.B. Digoxin um 60-90% (Verschraagen et al., 1999) oder Dabigatran um 100-150% steigert (Härtter et al., 2013). PGP wird nicht nur in Darmepithelien, sondern auch in der canaliculären Membran der Hepatozyten, in den proximalen Tubuluszellen der Niere und in den Endothelien der Hirnkapillaren exprimiert. Währendem der Effekt von Verapamil auf Dabigatran durch eine Steigerung der Bioverfügbarkeit erklärt werden kann, ist derjenige auf Digoxin vor allem eine Folge der gehemmten tubulären Sekretion.
Im Gegensatz zu den Dihydropyridinen sind Diltiazem und Verapamil negativ inotrop und hemmen die AV-Überleitung. Mit Digoxin und auch mit Betablockern, welche die AV-Überleitung ebenfalls hemmen, kommt es also zu einer pharmakodynamischen Interaktion, welche bei der Behandlung des tachykarden Vorhofflimmerns erwünscht sein kann, aber erkannt werden muss.
Thiazide
Als Antihypertensiva werden in der Schweiz vor allem Hydrochlorothiazid und Chlortalidon gebraucht. Beide haben eine gute Bioverfügbarkeit und werden via Filtration und Sekretion fast vollständig unverändert renal eliminiert.
Thiazide erhöhen die Lithiumkonzentration, was bei mit Lithium behandelten Patienten beachtet werden muss. Gleichzeitig verabreichte NSAIDs oder COX2-Hemmer verringern den antihypertensiven Effekt von Thiaziden, am ehesten wegen verstärkter Natriumretention unter NSAIDs/COX2-Hemmern. Die Kombination mit ACE-Hemmern oder ARBs ist beliebt; einerseits wegen des additiven antihypertensiven Effekts, andrerseits auch, weil die Tendenz der ACE-Hemmer und ARBs zu Hyperkaliämie durch die Thiazide ausgeglichen wird.
α2-Adrenozeptor-Agonisten
Als Antihypertensivum wird aus dieser Gruppe Clonidin verwendet. Clonidin hat eine gute Bioverfügbarkeit und wird zu ca. 50% unverändert renal ausgeschieden. Die Stimulation der präsynaptischen α2-Adrenozeptoren im Hirnstamm führt zu einer Reduktion des Sympathikus in der Peripherie und damit Blutdrucksenkung sowie zu Mundtrockenheit und Sedation als unerwünschte Wirkungen.
Wegen der Reduktion der Sympathikusaktivität verlangsamt Clonidin die AV-Überleitung, was bei einer Kombination mit Betablockern, Digoxin, Verapamil oder Diltiazem beachtet werden muss (Markowitz and Patrick, 2001). Der sedierende Effekt von Clonidin wird durch andere sedierende Medikamente und auch durch Alkohol verstärkt. α2-Adrenozeptorantagonisten wie z.B. Mirtazapin können den antihypertensiven Effekt von Clonidin vermindern oder aufheben. In der Literatur sind Fälle beschrieben, in denen es beim schnellen Absetzen von Clonidin insbesondere bei Patienten, welche gleichzeitig mit Propranolol behandelt werden, zu einem gefährlichen Anstieg des Blutdrucks kommen kann (Markowitz and Patrick, 2001). Reboundphänomene sind unter Clonidin gut bekannt, weshalb dieses generell langsam ausgeschlichen werden sollte. Da Propranolol die β2-Adrenozeptoren hemmt, welche eine Vasodilatation vermitteln, kann der Blutdruckanstieg nach schnellem Absetzen von Clonidin noch verstärkt werden.
α1-Adrenozeptor-Antagonisten
Doxazosin ist der in der Schweiz gebräuchliche Alphablocker. Doxazosin hat eine gute Bioverfügbarkeit (65%) und eine hohe Proteinbindung (98%) und wird vorwiegend renal und 95% metabolisiert ausgeschieden. CYP3A4 ist das hauptsächlich am Metabolismus von Doxazosin beteiligte Enzym.
Entsprechend steigt die Exposition von Doxazosin in Kombination mit CYP3A4 Inhibitoren. Diese Interaktion kann klinisch relevant sein, weil dosisabhängige unerwünschte Wirkungen von Doxazosin, insbesondere die Neigung zu Orthostase, verstärkt auftreten können. Im Gegensatz dazu können CYP3A4 Induktoren die Wirkung von Doxazosin abschwächen. Eine pharmakodynamische Interaktion ist das Auftreten von Hypotonien bei gleichzeitiger Einnahme von PD5-Inhibitoren wie z.B. Sildenafil. Mit Doxazosin behandelte Patienten sollten darauf hingewiesen werden, keine PD5-Inhibitoren einzunehmen.
Colchicin
Colchicin ist ein Alkaloid der Herbszeitlose. Die orale Bioverfügbarkeit beträgt wegen eines relevanten first-pass Effektes ca. 50%. Es ist zu 50% proteingebunden (first pass effect) und wird zu ca. 50% unverändert renal via Filtration und Sekretion ausgeschieden. 50% werden durch CYP3A4 metabolisiert (O-Demethylierung) und vorwiegend biliär eliminiert. Die Halbwertszeit liegt bei 15-30 Stunden, Colchicin unterliegt einem enterohepatischen Kreislauf. Colchicin ist, wie oben ausgeführt, ein Substrat von CYP3A4 und auch von PGP.
Die Interaktionen von Colchicin sind kürzlich sehr gut zusammengefasst worden (Hansten et al., 2023). Die pharmakokinetischen Interaktionen sind aufgrund der Substrateigenschaften von Colchicin voraussagbar. CYP3A4- and PGP-Hemmer steigern die Exposition von Colchicin und Induktoren fördern sie. Starke CYP3A4/PGP-Hemmer wie Clarithromycin, Imidazolantimykotika und Proteaseinhibitoren können den Colchicinspiegel bis 10-fach erhöhen und sollten deshalb vermieden. Mittelstarke CYP3A4/PGP-Hemmer wie Amiodaron, Diltiazem und Verapamil führen zu einer Verdoppelung bis Verdreifachung der Colchicinexposition, weshalb in diesem Fall die Dosierung von Colchicin um 50% gesenkt werden sollte. Cyclosporin ist ein mittelstarker bis starker CYP3A4/PGP-Hemmer und führt zu einer Verfünffachung der Colchicinexposition. Allerdings sind beide Substanzen myotoxisch, weshalb die Kombination Cyclosporin/Colchicin aus meiner Sicht kontraindiziert ist. Andere mit Myotoxizität assoziierte Arzneistoffgruppen wie z.B. Fibrate und Statine erhöhen das Risiko für Myotoxizität, aber nicht die Exposition von Colchicin. Falls Colchicin mit solchen Arzneistoffen kombiniert wird, sollten die Patienten auf das erhöhte Risiko für Myopathien hingewiesen und entsprechend überwacht werden.
Copyright bei Aerzteverlag medinfo AG
Klinische Pharmakologie & Toxikologie
Universitätsspital
4031 Basel
stephan.kraehenbuehl@usb.ch
Der Autor hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Marx N. et al., 2023 ESC Guidelines for the management of cardiovascular disease iBerger B, Donzelli M, Maseneni S, Boess F, Roth A, Krähenbühl S and Haschke M (2016) Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Front Pharmacol 7:443.
2. Dong H, Wang FZ, Shi K, Zhang XS and Lv DM (2021) Association of Cytochrome P450 2C9*3 and Angiotensin II Receptor 1 (1166A>C) Gene Polymorphisms With the Antihypertensive Effect of Irbesartan. Am J Hypertens 34:121.
3. Dresser GK, Bailey DG and Carruthers SG (2000) Grapefruit juice–felodipine interaction in the elderly. Clin Pharmacol Ther 68:28-34.
4. Egger SS, Meier S, Leu C, Christen S, Gratwohl A, Krähenbühl S and Haschke M (2010) Drug interactions and adverse events associated with antimycotic drugs used for invasive aspergillosis in hematopoietic SCT. Bone Marrow Transplant 45:1197-1203.
5. Hansten PD, Tan MS, Horn JR, Gomez-Lumbreras A, Villa-Zapata L, Boyce RD, Subbian V, Romero A, Gephart S and Malone DC (2023) Colchicine Drug Interaction Errors and Misunderstandings: Recommendations for Improved Evidence-Based Management. Drug Saf 46:223-242.
6. Härtter S, Sennewald R, Nehmiz G and Reilly P (2013) Oral bioavailability of dabigatran etexilate (Pradaxa(®) ) after co-medication with verapamil in healthy subjects. Br J Clin Pharmacol 75:1053-1062.
7. Hommers L, Fischer M, Reif-Leonhard C, Pfuhlmann B, Deckert J and Unterecker S (2019) The Combination of Lithium and ACE Inhibitors: Hazardous, Critical, Possible? Clin Drug Investig 39:485-489.
8. Kupferschmidt HH, Ha HR, Ziegler WH, Meier PJ and Krähenbühl S (1995) Interaction between grapefruit juice and midazolam in humans. Clin Pharmacol Ther 58:20-28.
9. Markowitz JS and Patrick KS (2001) Pharmacokinetic and pharmacodynamic drug interactions in the treatment of attention-deficit hyperactivity disorder. Clin Pharmacokinet 40:753-772.
10. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, Hess L, Schlienger RG and Krähenbühl S (2005) Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf 28:263-275.
11. Straubhaar B, Krähenbühl S and Schlienger RG (2006) The prevalence of potential drug-drug interactions in patients with heart failure at hospital discharge. Drug Saf 29:79-90.
12. Taavitsainen P, Kiukaanniemi K and Pelkonen O (2000) In vitro inhibition screening of human hepatic P450 enzymes by five angiotensin-II receptor antagonists. Eur J Clin Pharmacol 56:135-140.
13. Tamblyn RM, McLeod PJ, Abrahamowicz M and Laprise R (1996) Do too many cooks spoil the broth? Multiple physician involvement in medical management of elderly patients and potentially inappropriate drug combinations. Cmaj 154:1177-1184.
14. Verschraagen M, Koks CH, Schellens JH and Beijnen JH (1999) P-glycoprotein system as a determinant of drug interactions: the case of digoxin-verapamil. Pharmacol Res 40:301-306.
15. Vonbach P, Dubied A, Krähenbühl S and Beer JH (2008) Prevalence of drug-drug interactions at hospital entry and during hospital stay of patients in internal medicine. Eur J Intern Med 19:413-420.
16. Waldorff S, Hansen PB, Egeblad H, Berning J, Buch J, Kjaergård H and Steiness E (1983) Interactions between digoxin and potassium-sparing diuretics. Clin Pharmacol Ther 33:418-423.

der informierte @rzt
- Vol. 14
- Ausgabe 2
- Februar 2024