

In den letzten Jahren ist eine deutliche Zunahme der Sichelzellerkrankung in unseren Breitengraden zu verzeichnen. Grund dafür sind die Flüchtlingsströme der letzten Jahre, wobei Menschen mit Sichelzellanämie aus dem Nahen Osten und Nordafrika nach Europa gekommen sind und noch immer kommen. Nicht nur die Hämatologen, sondern auch die Kollegen der Notfallmedizin sind gefordert. Eine Übersicht über die akuten und chronischen Komplikationen dieser für uns seltenen Krankheit und deren Therapie sind hier dargestellt.
Im Englischen wird die Sichelzellerkrankung mit dem Überbegriff „sickle cell disease“ bezeichnet. Dabei werden genetisch unterschiedliche Krankheiten zusammengefasst, die sich klinisch ähnlich äussern. Weitaus am häufigsten ist die homozygote Mutation in der Betaglobulinkette des Hämoglobins (Hb), wobei an der Position 6 ein Valin durch ein Glutamin ersetzt wird (Genotyp HbSS). Als Variante kann auch ein Gemisch aus einer heterozygoten Sichelbetaglobulinmutation mit einer anderen Betaglobulinmutation vorliegen. Erwähnenswert ist zudem die verhältnismässig häufige Kombination mit einer Thalassämie. Dabei ist die Sichelzell-Beta-Thalassämie klinisch am relevantesten. Je nachdem ob noch eine Restproduktion von Betaglobulinketten vorliegt, kann dabei zwischen Sichelzell-Beta+ Thalassämie (gehäuft bei Afro-Amerikanern) oder Sichelzell-Beta0 Thalassämie (gehäuft in Griechenland, Mittelmeerregion und mittlerem Osten) unterschieden werden.
Normales deoxygeniertes Hb ist im Erythrozyten (EC) gut löslich. Deoxygeniertes HbS jedoch agglutiniert schnell. Dies führt, zusammen mit einer Veränderung der Struktur und Funktion der EC-Membran sowie einer gestörten Kontrolle des Zellvolumens, zur Sichelform der EC. Die Sichelzellen weisen zudem eine erhöhte Adhärenz an Gefässendothelien auf.
Von der Sichelzellerkrankung heterozygot Betroffene entwickelten über die Jahrmillionen einen Selektionsvorteil. Die Anfälligkeit für Malaria ist geringer, da die Plasmodien in den Sichelzellen weniger lange überleben.
Diagnose
Wenn auch selten, so finden sich klassischerweise im mikroskopischen Ausstrich die sichelförmig verzogenen EC, die Sichelzellen. Daneben sind eine Polychromasie und Howell Jolly Körperchen als Zeichen einer funktionellen Asplenie häufig. Mittels Analyse des HbS (zum Beispiel Hb-Elektrophorese) kann die Diagnose mit Sicherheit gestellt werden.
Klinik
Für die Klinik der Sichelzellerkrankung hauptverantwortlich sind folgende zwei Pathomechanismen: einerseits eine Hämolyse, andererseits Gefässverschlüsse durch Agglutinieren von ECs. Daraus lassen sich in der Folge die akuten und chronischen Komplikationen der Erkrankung herleiten.
Akute Komplikationen
Eine chronisch kompensierte hämolytische Anämie kann sich akut verschlechtern. Ursachen hierfür können eine aplastische Krise (zum Beispiel durch einen Infekt mit Parvovirus B19), Hyperhämolyse oder Milzsequestration sein.
Starke Schmerzen als Folge eines Gefässverschlusses sind für die Sichelpatienten klinisch mit am belastendsten. Sind beispielsweise die Mesenterialgefässe verschlossen, kommt es als Folge der Infarzierung zu einem paralytischen Ileus (auch Girdle Syndrom genannt). Prinzipiell können durch die Gefässverschlüsse alle Organsysteme betroffen sein, so dass es zu ophthalmologischen Syndromen (Retinalarterienokklusion, Glaskörpereinblutung, Amotio), Niereninfarkten, akuten neurologischen Komplikationen (das Risiko für einen Krampfanfall ist 3 x häufiger als in der Normalbevölkerung), Priapismus, Schwangerschaftskomplikationen und venösen Thromboembolien kommen kann.
Ein besonderes Augenmerk ist auf die folgenden beiden akuten Komplikationen zu legen:
1. Akutes Thoraxsyndrom, englisch Acute Chest Syndrome (ACS). Die Mortalität des ACS beträgt bei den Erwachsenen bis 10%. 50% aller Sichelzellpatienten erleiden in ihrem Leben ein ACS. Es tritt häufig ohne ersichtlichen Grund auf, kann aber mit einem viralen Infekt oder lokalen Traumata assoziiert sein (Tabelle 1). Differenzialdiagnostisch kommen für das ACS primär Lungenembolien, eine Pneumonie oder ein akutes Koronarsyndrom in Frage. Während Lungenembolien und Pneumonien sehr häufig sind, erleiden Sichelzellpatienten bezogen zur Normalbevölkerung nur selten einen Herzinfarkt. Grund dafür ist ein optimaleres Lipidprofil mit tieferen Werten von LDL und totalem Cholesterin (1).
2. Infekte. Diese sind nebst ACS Hauptursache für die Morbi-
dität und Mortalität der Sichelzellerkrankung. Das Leitsymptom ist dabei Fieber. Allerdings kann Fieber auch bei anderen sichelzelltypischen Erkrankungen wie dem ACS auftreten. Zu den häufigsten nachgewiesenen Erregern zählen Strept. pneumoniae, Salmonella spp., Hämophilus influenzae Typ b, E. coli, Staph. aureus. Insbesondere ist das Infektrisiko auf Grund einer funktionellen Asplenie (siehe unten) auch für andere bekapselte Erreger wie zum Beispiel Meningokokken erhöht.
Chronische Komplikationen
Unter der repetitiven Vasookklusion leiden alle Organe. Über die Jahre kann es zu kognitiven Einschränkungen, poliferativer Retinopathie, Kardiomyopathie, pulmonaler Hypertonie, Niereninsuffizienz, Cholezystolithiasis, Osteonekrose, Beinulcera, erektiler Dysfunktion, verzögerter Pubertät und Kleinwuchs kommen.
Die wichtigste Langzeitkomplikation ist eine funktionelle Hypo- bis Asplenie. Dies manifestiert sich bereits in jungen Jahren und zieht ein erhöhtes Infektrisiko mit sich.

Management und Therpie
Die Therapie der Sichelzellerkrankung ist vielschichtig:
Prävention:
1. Screening: Pränatal erfolgt das Screening mittels DNA-PCR aus einer Chorionzottenbiopsie. Eine liquid biopsy bei der Mutter hat aktuell (noch) keinen Stellenwert. Weil das HbS erst in den ersten Lebenswochen durch sukzessiven Ersatz des HbF entsteht, liefert die Hb-Elektrophorese beim Neugeborenen falsch-negative Resultate.
2. Schulung der Patienten: Beispielsweise sollten sich Sichelzellpatienten bei Fieber über 38.5°Celsius sofort in ärztliche Behandlung begeben.
3. Impfungen: Gegen Erreger, gegen welche Impfungen vorhanden sind, sollte geimpft werden (Pneumokokken, H. influenzae, Meningokokken und Influenza).
4. Penicillinprophylaxe: Bereits kleinste Kinder können eine funktionelle Asplenie haben, weswegen ale Kinder unter 5 Jahren eine Penicillinprophylaxe erhalten sollten.
Sichelzellspezifische Therapie
1. Management einer Schmerzkrise
Analgetika. Leichte Schmerzen können mit Paracetamol, nicht-steroidalen Antirheumatika oder Tramadol kontrolliert werden. Bei starken Schmerzen empfiehlt sich ein Opiat, wobei die erste Wahl Morphium und bei eingeschränkter Nierenfunktion Hdromorphon ist.
Sauerstoff. Die periphere Sauerstoffsättigung sollte wenn möglich über 95% liegen.
Flüssigkeit. Die Patienten sind in der Regel auf Grund verminderter Flüssigkeitsaufnahme und veränderter Konzentrierungsmöglichkeit der Niere hypovoläm. Das Ziel ist eine leicht positive Flüssigkeitsbilanz bei stabilem Gewicht.
Thromboseprophylaxe. Zur Vermeidung einer Thrombose wird am besten niedermolekulares Heparin eingesetzt.
2. Transfusion
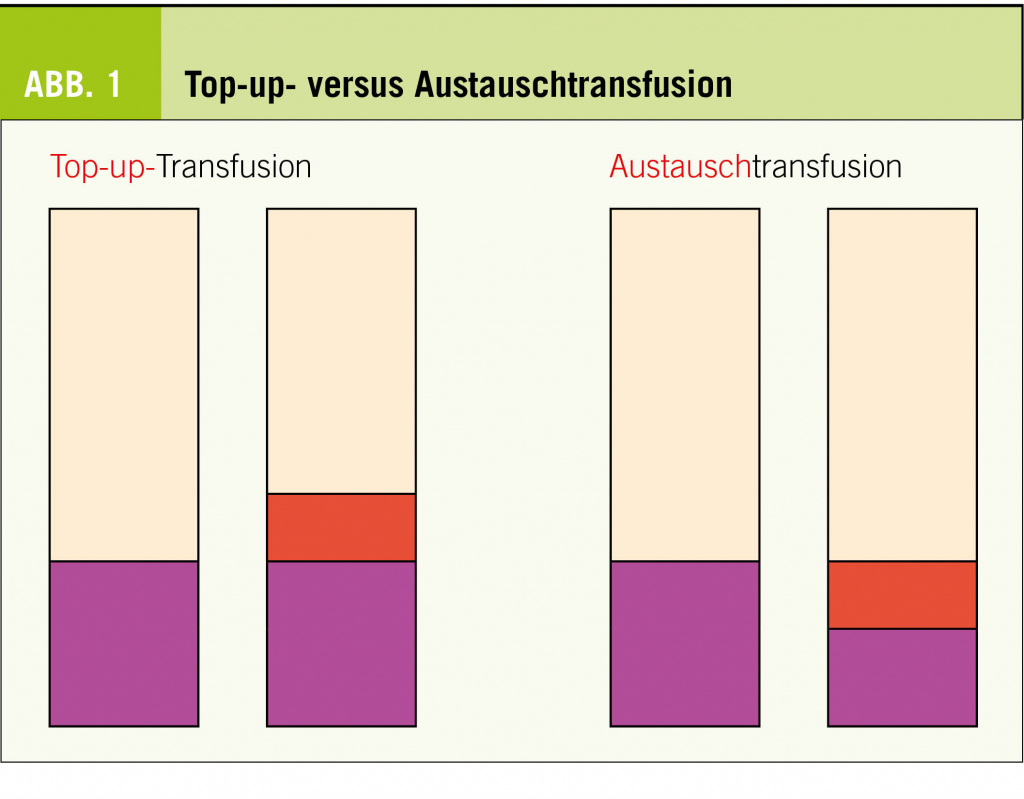
EC-Transfusionen sind einerseits Prophylaxe, andererseits Therapie einer akuten Vasookklusion wie Insult oder ACS. Transfusionen heben den Hämatokrit und mindern den prozentualen Anteil vom HbS (Ziel wäre < 30%). was beides die Wahrscheinlichkeit einer Vasookklusion senkt. Durch die Transfusionen wird die Produktion von Erythropoietin vermindert, womit folglich weniger HbS produziert wird. Allerdings besteht die Gefahr einer Alloimmunisierung sowie Eisenüberladung. Es kann zwischen Top-up-und Austauschtransfusion unterschieden werden, wobei die Indikationen unterschielich sind (Abb. 1).
3. Hydroxyurea (HU)
Seit Jahrzehnten wird dieses Medikament in der Therapie der myeloproliferativen Erkrankungen eingesetzt. Nebst der Beeinflussung der Zellteilung durch Hemmer der Ribonukleotidreduktase bewirkt HU einen Shift in der Genexpression. Dadurch kommt es zu einer vermehrten Produktion von HbF und die relative Konzentration von HbS sinkt. Der Wirkmechanismus ist bei der Sichelzellerkrankung allerdings noch nicht zu 100% verstanden.
Organspezifische Therapie
Chronisch geschädigte Organe sollten best möglichst unterstützt werden, beispielsweise bei einer Niereninsuffizienz mit ACE-Hemmern.
Kuratio
Die einzige aktuell verfügbare kurative Therapie ist die hämatopoietische Stammzelltransplantation. Die besten Daten hierfür liegen für HLA-identische Familienspender vor (2). Weil durch die Sichelzellerkrankung schon früh viele Organe geschädigt sein können und eine Transplantation damit schlechter toleriert wird, sollte diese möglichst vor dem Alter von 16 durchgeführt werden.
Experimentelle Therapie
1. Medikamente, die verhindern, dass Erythrozyten am Gefässendothel adhärieren, sind in Entwicklung. Ein Beispiel ist Crizanlizumab. Dieser P-Selectin-Antikörper wird aktuell in einer Phase 3 Studie geprüft, nachdem in einer Phase 2 Studie eine signifikante Verminderung von Schmerzkrisen gezeigt werden konnte (3).
2. Antioxidantien wie L-Glutamin. Auch dieses Medikament führte als Monotherapie oder in Kombination mit Hydroxyurea in einer kürzlich publizierten Studie zu einer Reduktion von Schmerzkrisen (4).
3. Schlussendlich stellt die Sichelzellerkrankung mit ihrerdefinierten genetischen Mutation prinzipiell eine ideale Erkrankung für die Gentherapie dar. Allerdings liegt die Anwendbarkeit für die Klinik noch in weiter Ferne.
Onkologie KSGR
Loëstrasse 170
7000 Chur
katharina.huss@ksgr.ch
Die Autorin hat keine Interessenskonflikte in Zusammenhang mit diesem Beitrag.
Literatur:
1. El-Hazmi MA. Cholesterol and triglyceride level in patients with sickle cell anaemia. Scand J Clin Lab Invest. 1987 Jun;47(4):351-4.
2. Gluckman E. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017 Mar 16;129(11):1548-1556.
3. Ataga K.I. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017;376:429-439.
4. Yutaka N. A phase 3 trial of L-Glutamine in sickle cell disease. N Engl J Med 2018;379:226-235.

der informierte @rzt
- Vol. 8
- Ausgabe 9
- September 2018