
Thalassämien gehören zu den häufigsten monogenen Erbkrankheiten weltweit. Die Labordiagnostik der Thalassämien erfolgt in zwei Stufen, erstens mit dem spezifischen «Hämoglobin-Screening» und zweitens mit der Ergänzung und Bestätigung durch «Molekulardiagnostik» einschliesslich genetischer Beratung. Zielpopulation der Thalassämiediagnostik sind Patienten mit Mikrozytose (MCV<80 fL) oder Hypochromie (MCH<27pg) unklarer Ätiologie, Alters unabhängig und mit erhöhtem Risiko für Hemoglobinopathien im Hintergrund (familiäre Belastung mit chronischer Anämie oder Ethnie wegen der endemischen Gebiete oder abnorme Laborparameter). Die Abklärung ist besonders für Paare präkonzeptionell wichtig, wegen der Familienplanung. Eine spezialisierte genetische Beratung ist hier erforderlich.
Thalassemias are among the most common monogenic inherited diseases worldwide. Laboratory diagnosis of thalassemias is performed in two stages, first with the targeted “hemoglobin screening” and second with supplementation and confirmation by “molecular diagnostics” including genetic counseling. The candidate population for thalassemia diagnostics are patients of all ages with microcytosis (MCV<80 fL) or hypochromia (MCH<27pg) of unknown etiology with increased risk for thalassemia in the background (family history of chronic anemia or ethnicity due to the endemic areas for hemoglobinopathies or abnormal laboratory findings). Investigation is especially important for couples at preconception, because of the family planning. Specialized genetic counseling is required here.
Key Words: Thalassämieabklärung, Hämoglobinopathien, Mikrozytose, Hypochromie, Molekulargenetik
Thalassämien gehören zu den häufigsten Erbkrankheiten weltweit. Endemisch betroffen sind traditionellerweise Länder des Mittelmeers, Südostasien und Nordafrika. Die Vereinfachung und Globalisierung der Völker-Migration jedoch, hat dazu geführt, dass diese klinischen Entitäten zum Alltag der medizinischen Versorgung gehören auch in Ländern, wo dies früher eine Seltenheit war. Die Komplexität und Diversität der Diagnose und Therapie dieser Syndrome erfordern eine erhöhte Sensibilisierung der Gesundheitsanbieter. Die technologische Entwicklung der diagnostischen Möglichkeiten der Labordiagnostik erlaubt uns heute eine effiziente, zielgerichtete und ökonomische Diagnose. Im Folgenden werden wir diese Möglichkeiten erläutern.
Pathophysiologie
Bei gesunden Personen bildet das zirkulierende Hämoglobin keine einheitliche Fraktion; vielmehr lassen sich drei verschiedene Hämoglobin-Fraktionen feststellen, die sich nicht nur in der Konzentration, sondern auch in der Struktur der Globinkette unterscheiden (Tab. 1).

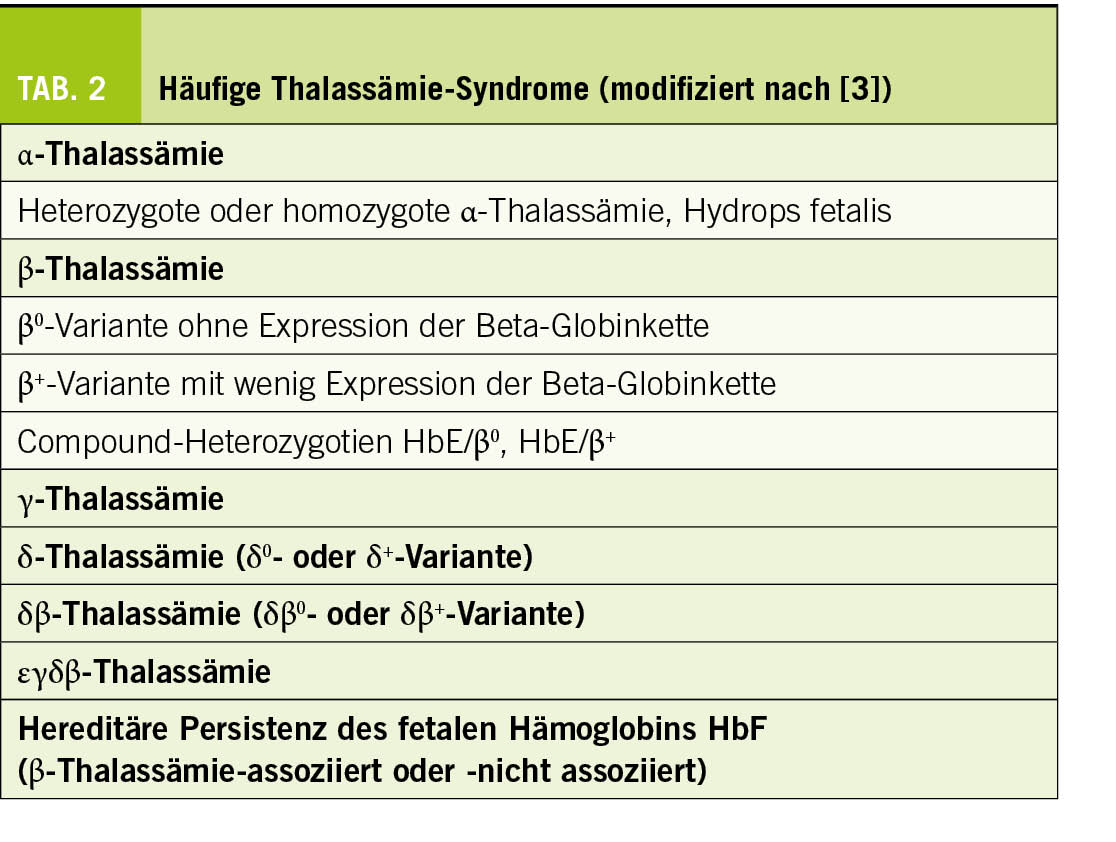
Thalassämien sind monogene Erbkrankheiten. Mehrere hunderte verschiedene Mutationen der Gene der Globinketten sind bisher als Ursache beschrieben (Tab. 2). Dieser genetische Hintergrund führt zu einem gestörten Gleichgewicht der Produktion der Globinketten, und nachfolgend zur intramedullären Apoptose der Vorläuferformen der erythroiden Zelllinie und zur ineffektiven Erythropoese. Klinisch resultiert dies in einem breiten Bild von der stummen Trägerform bis zur transfusionsbedürftigen Anämie. Die frühzeitige effiziente Diagnose und genetische Beratung tragen im Sinne der Prävention dazu bei, die mittelschweren und schweren klinischen Bildern zu vermeiden.
Diagnostische Möglichkeiten
Die Labordiagnostik von Thalassämien und Hämoglobinopathien generell, erfolgt in zwei verschiedene Ebenen: 1. In der ersten Stufe mit dem sogenannten «Hämoglobin-Screening». 2. In der zweiten Stufe mit der Ergänzung und Bestätigung durch «Molekulardiagnostik» (einschliesslich genetischer Beratung).
Zielpopulation für die Thalassämiediagnostik
A. Patienten mit Mikrozytose unklarer Ätiologie (MCV<80 fL) mit oder ohne Anämie (MCH<27pg) und Patienten mit erhöhtem Risiko für Thalassämie (Familiärer Belastung mit chronischer Anämie, Ethnie bezüglich endemischer Gebiete, abnorme Laborbefunde). Diese Abklärung ist besonders für Paare präkonzeptionell wichtig, wegen der Familienplanung. Eine spezialisierte genetische Beratung ist hier erforderlich. Als Möglichkeit bzw. als Teil dieser Beratung steht auch die genetische Präimplantationsdiagnostik zur Verfügung.
B. Schwangere mit erhöhtem Risiko für Thalassämie, basierend auf Anamnese, oder familiärer Belastung mit Anämie, oder Ethnie (endemische Gebiete für Thalassämie), oder Laborbefunde (MCV<80 fL oder MCH<27 pg). Oft hat man hier zeitlich gesehen keinen Spielraum für den konventionellen Ausschluss von Differentialdiagnosen, z.B. eines Eisenmangels, daher ist der Sprung in die effizientere Molekulardiagnostik unerlässlich.
C. Neugeborene mit erhöhtem Risiko für Thalassämie, je nach geographischer Lokalisation (endemische Gebiete) sind nationale, fokussierte Screening-Programme in Kraft. In der Schweiz ist das Neugeborenen-Screening für Hämoglobinopathien nicht obligatorisch.
1. Hämoglobin-Screening
Dieses beinhaltet ein komplettes alterskorreliertes Hämatogramm mit Ermittlung aller Erythrozyten-Indizes, sowie die spezifische Methode HPLC (Hochleistungs-Flüssigchromatographie, «High Performance Liquid Chromatography»), welche die verschiedenen normalen und/oder abnormen Hämoglobin-Varianten quantitativ abbildet. Die wichtigsten Hämoglobin-Fraktionen einer Probe werden im Chromatographieverfahren anhand ihrer spezifischen Retentionszeit-«Fenster» chemisch-analytisch identifiziert und quantifiziert.
In etwa 90 % der Fälle kann damit eine zutreffende, korrekte Diagnose gestellt werden, bei «klassischen» Beta-Thalassämie-Trägern und/oder Sichelzellanomalie (mit Einbindung des Sichelzelltests) sogar in 100 % der Fälle. Bei den beiden letztgenannten Erkrankungen handelt es sich um die bedeutendsten beta-Globin-Gendefekte – sowohl in Bezug auf die gesundheitlichen Auswirkungen im Einzelfall als auch im Hinblick auf die Bewertung des Risikos von Thalassämie- und/oder Sichelzell-Syndromen im Rahmen der Beratung von Paaren mit Kinderwunsch.
In einigen Fällen, z. B. milde und schwere Formen der Alpha-Thalassämie, kann die Diagnose nur genetisch gestellt werden, und soll daher gemäss den einschlägigen Leitlinien grundsätzlich durch molekulargenetische Untersuchungen bestätigt werden.
2. Molekulardiagnostik
Die Molekulardiagnostik umfasst definitionsgemäss die diagnostischen Abklärungen der zweiten Stufe, die im Wesentlichen der Vervollständigung und Bestätigung der Ergebnisse aus dem Hämoglobin-Screening dienen.
Zwei der wichtigsten Techniken sind die DNA-Sequenzierung (Bestimmung der Basenabfolge oder NGS «Next Generation Sequencing») zur Mutationsanalyse sowie die multiplexe ligationsabhängige Sondenamplifikation (MLPA) zur Erkennung grosser Deletionen in den Gendefekten.
Bei der Durchführung von molekulargenetischen Untersuchungen sind einige Empfehlungen zu beachten: Vor der DNA-Analyse ist eine hämatologische Bewertung des Falles wichtig für die Wahl der am besten geeigneten molekulargenetischen Methode, und nach der DNA-Analyse ist eine hämatologische Bewertung der genotypisch-phänotypischen Korrelation der Ergebnisse obligatorisch, um eine Fehldiagnose auszuschliessen (Tab. 3).

Die korrekte Charakterisierung der Träger-Genotypen bildet die Grundlage der adäquaten genetischen Beratung und ist eine zentrale Voraussetzung für die pränatale Diagnostik.
Auch bei grenzwertigen HPLC-Resultaten der ersten Stufe (z.B, grenzwertig normales HbA2), lohnt es sich gezielt molekulargenetisch die Diagnostik fortzusetzten. Dabei können stumme Formen der Alpha- oder Beta-Thalassämiesyndrome («silent carrier») aufgedeckt werden (in endemischen Gebieten 1%-8% der Verdachtsdiagnosen). Diese hätte man sonst verpasst, weil die Erythrozytenindizes dabei nicht auffällig werden. Zwar hätten diese «stummen» Formen phänotypisch für die betroffene Person keine relevanten klinischen Konsequenzen, wären aber von grosser Relevanz im Rahmen einer Familienplanung und Familienberatung.
Copyright bei Aerzteverlag medinfo AG
SYNLAB Suisse SA
Alpenquai 14
6002 Luzern
dimitrios.tsakiris@usb.ch
SYNLAB Suisse SA
Via Pianon 7
6934 Bioggio
SYNLAB Suisse SA
Via Pianon 7
6934 Bioggio
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Traeger-Synodinos J, Harteveld CL, Old JM, et al. EMQN best practice guidelines for molecular and haematology methods for carrier identification and prenatal diagnosis of the haemoglobinopathies. Eur J Hum Genet 2015; 23 (4): 426-437 doi: 10.1038/ejhg.2014.131
2. Mettananda S, Gibbons RJ, HiggsDR.α-Globin as a molecular target in the treatment of β-thalassemia. Blood 2015; 125 (24): 3694-3701 doi: 10.1182/blood-2015-03-633594
3. Mensah C, Sheth S. Optimal strategies for carrier screening and prenatal diagnosis of α- and β-thalassemia. Hematology Am Soc Hematol Educ Program. 2021 Dec 10;2021(1):607-613 doi: 10.1182/hematology.2021000296
4. Ryan K, Bain BJ, Worthington D, et al. Significant haemoglobinopathies: guidelines for screening and diagnosis. Br J Haematol 2010; 149: 35-49 doi: 10.1111/j.1365-2141.2009.08054.x
5. Colaco S, Colah R, Nadkarni A. Significance of borderline HbA2 levels in β thalassemia carrier screening. Sci Rep. 2022; 12(1):5414. doi: 10.1038/s41598-022-09250-5



der informierte @rzt
- Vol. 13
- Ausgabe 5
- Mai 2023