

Die Vorgänge der Blutgerinnung werden durch ein faszinierendes Wechselspiel zwischen Zahl und Funktion der Blutplättchen und der plasmatischen Gerinnungskaskade bestimmt. Blutungsbilder in der Praxis sind häufig. Thrombozytopenien können häufig erworben oder selten angeboren sein, sich über Monate abzeichnen oder aber praktisch über Nacht zutage treten. Ein präanalytisches Problem soll neben den klinischen differentialdiagnostischen Überlegungen immer in Erwägung gezogen werden. In jedem Fall gilt es, sich eine Übersicht des gesamten Blutbildes zu verschaffen. Der Ort der Blutung (Haut, Schleimhaut), der auslösende Faktor (inadäquates Trauma oder spontan), die Form der Blutungsmanifestation (Petechien als Zeichen) und der Zeitfaktor (pathologisch verlängerte Blutung, Alter) lassen an eine thrombozytopenische Blutung denken.
The processes of blood coagulation are determined by a fascinating interplay between the number and function of platelets and the plasmatic coagulation cascade. Thrombocytopenias may be often acquired or in rare cases congenital, may become apparent over months, or may appear virtually overnight. A preanalytical problem should always be considered as a causative factor beside the clinical differential diagnosis. In any case, it is important to obtain an overview of the complete blood count and morphology. Bleeding patterns in practice are common. Site of bleeding (skin, mucosa), precipitating factor (inadequate trauma or spontaneous), form and signs of bleeding (purpura) and time factor (pathologically prolonged bleeding, age) suggest thrombocytopenic bleeding.
Key Words: Thrombocytopenia, autoimmune thrombocytopenia, congenital thrombocytopenia, hemostasis disorder, acquired and congenital bleeding tendency.
Erworbene Thrombozytopenien
Pseudothrombozytopenie / Satellitenbildung
Thrombozyten neigen in vitro dazu, Aggregate zu bilden. Begünstigend auf dieses Phänomen wirken Aktivierung der Plättchen durch traumatische Venenpunktionen, wobei auch die Art des Antikoagulans, mit dem das Blutröhrchen versehen ist, von Bedeutung ist. Plättchenklumpen können bei sämtlichen Monovetten vorkommen, sind im Citratblut aber um ein Vielfaches seltener als im üblicherweise für Blutbilder verwendeten EDTA-Röhrchen. Dieses Verklumpen führt zu falsch tiefen Plättchenzahlen in automatisierten Analysegeräten und geschieht unabhängig von dem gewählten Hämatologie-Analysesystem: Im Impedanzmessprinzip, wie es in den meisten Praxen Verwendung findet, werden Blutbestandteile in einer Volumenverteilungskurve aufgetragen, mit dem Nachteil, dass besonders grosse oder eben verklumpte Thrombozyten fälschlicherweise als kleine Erythrozyten oder als Lymphozyten registriert werden. In der hydrodynamischen Fokussierung mit Durchflusszytometrie (grosse Blutbildautomaten), wie sie in Spitälern angewendet werden, finden sich zwar integrierte Regelwerke, die auf eine mögliche Pseudothrombopenie durch Verklumpung hinweisen, aber der verlässlichste Weg, den Verdacht zu bestätigen, ist und bleibt die mikroskopische Beurteilung.
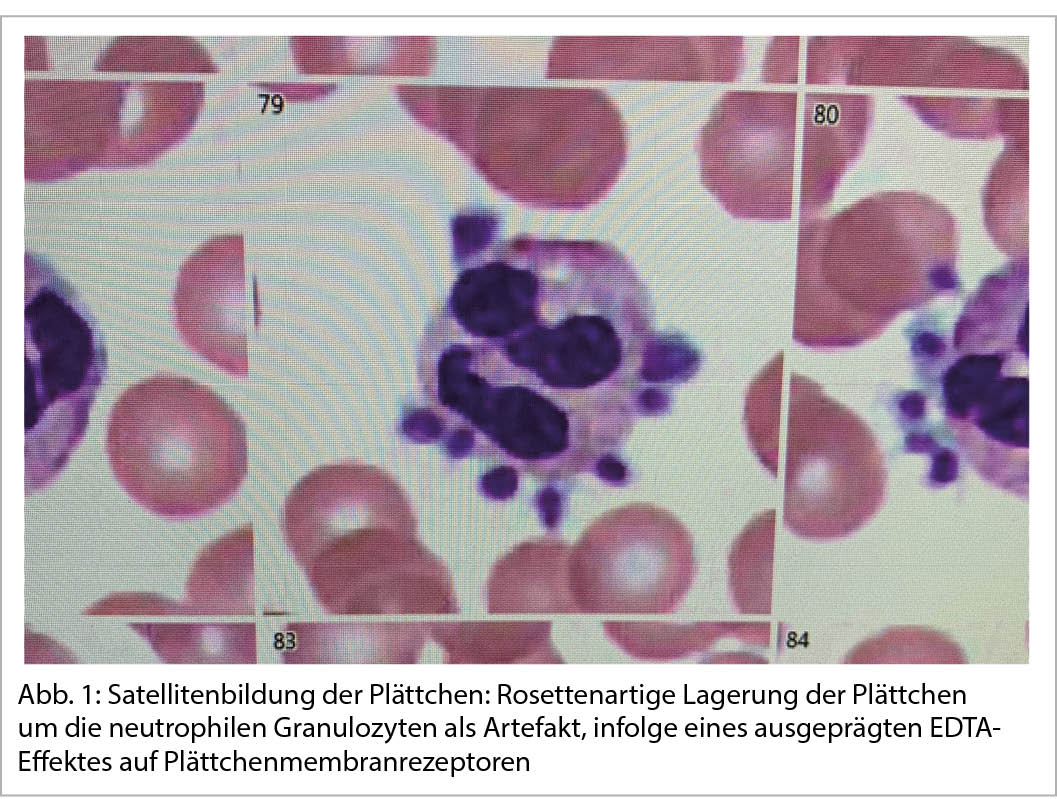
Die Satellitenbildung ist ein weiteres In-vitro-Phänomen, wie es hauptsächlich im EDTA-Blut vorkommt: Rosettenartig lagern sich dabei die Plättchen an die Membran der Neutrophilen (Abb. 1) mit folglich falsch tiefen Thrombozytenwerten.
Beide Phänomene (Pseudothrombozytopenie und/oder Satellitenbildung) beruhen auf folgendem Mechanismus. EDTA als Kalzium-Chelator entnimmt Kalzium-Ionen aus der tertiären Struktur des Fibrinogenrezeptors GPIIb/IIIa auf der Plättchenmembran. Dies verändert die Symmetrie des Rezeptors so weit, dass ein kryptisches Epitop aufgedeckt wird. Dieses wird von zufälligen sonst nicht pathogenen Autoantikörpern (IgG-Moleküle) erkannt und gebunden, was zur Aneinanderhaftung der Plättchen (Plättchenaggregate) oder zu den Neutrophilen (Satellitenbildung) führt.
Autoimmunthrombopenie (ITP)
Die Immunthrombozytopenie, ITP auch Morbus Werlhof, wird im Kindesalter oft als vorübergehende Begleiterscheinung eines Infekts beobachtet. Mit zunehmendem Alter werden chronische Verlaufsformen häufiger (Prävalenz 1:10 000). Ursächlich zeichnen eine Vielzahl immunologischer Dysregulationen verantwortlich für diese Form des Plättchenmangels. Es handelt sich in der Regel um eine isolierte, nicht selten mittelschwer- bis schwergradige Thrombozytopenie, die durchaus mit Blutungskomplikationen einhergehen kann. Die ITP ist praktisch eine Ausschlussdiagnose und wird meistens erfolgreich medikamentös behandelt. Es haben sich diverse Neuerungen im Bereich der medikamentösen Therapie (Steroid-Stoss-Therapie, Thrombopoietin-Rezeptor-Agonisten) durchgesetzt, so dass eine Splenektomie als Behandlung sich nur in äusserst refraktären Fällen anbietet.
Medikamentenbedingte Thrombozytopenien
Zahlreiche Medikamente wurden als Auslöser einer Thrombozytopenie beschrieben. Am häufigsten wurden Reaktionen mit Chinidin, Chinin-haltigen Produkten, Penicillinen, Ranitidin, Methyldopa, Procainamid berichtet. Das Medikamentenmolekül wird durch Opsonisierung zum Antigen und ruft Antikörper hervor. Letztere erkennen zufällig Epitope auf den Membranrezeptoren der Plättchen, welche dann als «innocent bystanders» in der Peripherie schneller eliminiert werden. Das Absetzen des Medikamentes korrigiert die Thrombozytopenie und die Wiederaufnahme führt zu einem Rezidiv.
Die Heparin-induzierte Thrombozytopenie (HIT) ist nach wie vor ein häufig anzutreffendes Phänomen im Klinikalltag und wird Antikörper-mediiert hervorgerufen, in der Regel durch das dort geläufigere unfraktionierte Heparin. Seltener, sind es niedermolekularen Heparine, die eine HIT auslösen können. Diagnostisch wird als erstes der spezifische 4T-Score für die Schätzung der klinischen Vortestprobalilität errechnet. Labortechnisch wird die Diagnose durch den Nachweis von anti-PF4/Heparin Antikörpern bestätigt. Therapeutisch wird auf alternative Antikoagulanzien wie Argatroban (Argatra®), danaparoid (Orgaran®), Fondaparinux (Arixtra®) oder direkte orale Antikoagulanzien (DOAC) umgestellt.
Andere Ursachen
Weitere Formen erworbener Thrombozytopenien stellen der Hypersplenismus bei Leberzirrhose, die Schwangerschaftsthrombopenie, die disseminierte intravasale Gerinnung (DIC), die mikroangiopathischen Thrombozytopenien (thrombotisch thrombozytopenische Purpura TTP, hämolytisch urämisches Syndrom HUS), wie auch maligne hämatologische Erkrankungen und die Tumor-spezifischen Chemotherapien dar (Tab. 1). Bei den mikroangiopathischen Thrombozytopenien besitzt die morphologische Erkennung der Fragmentozyten oder Schistozyten in der mikroskopischen Beurteilung des peripheren Blutausstriches grossen diagnostischen Wert.

Hereditäre Thrombozytopenien
Das Erkennen angeborener Formen der Thrombozytopenie ist von grosser Wichtigkeit, zum einen, um korrekte therapeutische Massnahmen (z.B. perioperative Gerinnungskontrolle) zu definieren, zum anderen, um die Betroffenen vor unnötigen diagnostischen Schritten oder unsachgemässen Therapieversuchen zu bewahren: Fälle von als «einfache» Immunthrombopenie verkannten angeborenen Thrombozytopenien mit folglich langjähriger Gabe immunsupressiver Agenzien bis hin zur infausten Splenektomie sind beschrieben (Tab. 1).
Makrothrombozytopenien
Bei der sogenannten Makrothrombozytopenie kann bereits aufgrund der mikroskopischen Analyse eines Blutausstrichs der Verdacht darauf geäussert werden: diverse Gendefekte im MYH9 Gen (Myosin-Gen, in den meisten Fällen autosomal dominant vererbt) führen zu verfrühtem Übertritt der Thrombozyten vom Knochenmark ins periphere Blut, mit sichtbaren Riesenthrombozyten und gegebenenfalls charakteristischen zytoplasmatischen Inklusionen in den Leukozyten. Im maschinellen Blutbild fällt das pathologisch erhöhte mittlere Plättchenvolumen (MPV) auf.
Zu den MYH9-assoziierten Makrothrombozytopenien gehören die May-Hegglin-Anomalie mit charakteristischen zytoplasmatischen Inklusionskörperchen in den Neutrophilen (Döhle Bodies), das Epstein-Syndrom, das sich mit Innenohrschwerhörigkeit und Nephritis klinisch äussert, das Fechtner-Syndrom, mit zusätzlicher Neigung zu präsenilem Katarakt, und das Sebastian-Syndrom. In den genannten Formen erreicht die Thrombozytenzahl in der Regel Werte zwischen 30 und 100 G/L. Klinisch dominieren entsprechend eher Blutergüsse, Ekchymosen und übermässige Regelblutungen.
Petechien sind untypisch. Nicht selten sind die Indexpatientinnen einer Familie mit hereditärer Makrothrombozytopenie junge Frauen in Abklärung eines Eisenmangels. Die vermehrte Blutungsneigung resultiert aus einem instabilen Plättchenthrombus. Der unauffällige zytomorphologische und histopathologische Befund im Knochenmark (durchreifende, allenfalls leicht gesteigerte Megakaryopoiese) ist mitunter Grund für Fehldiagnosen wie die der Immunthrombozytopenie. Eine Knochenmarkpunktion ist entsprechend nicht zielführend. Die Mikroskopie des peripheren Blutausstrichs ist umso mehr ein unerlässliches Mittel zum Screening. Zudem geben moderne Blutbildgeräte das mittlere thrombozytäre Volumen (MPV) erhöht an. In manchen Geräten werden die grossen Plättchen fälschlicherweise den Erythrozyten oder den Leukozyten zugeordnet. Plättchenfunktionstests (Plättchenaggregation nach Born), wie sie an grösseren Laboratorien angeboten werden, zeigen keine Auffälligkeiten bei MYH9-assoziierten Makrothrombozytopenien.
Bernard Soulier Syndrom (BSS)
Auf andere Weise kommt es beim BSS zu einer Blutungsneigung: Durch den fehlenden Glykoprotein Ib/IX-Rezeptor auf den Plättchen sind diese nicht dazu in der Lage, über den von Willebrand Faktor an das verletzte Gefässendothel anzuheften (Adhäsion). Die Plättchengrösse kann erhöht sein, die Thrombozytopenie ist aber eher mild mit 100 bis 120 G/L. Plättchenfunktionstests erlauben die Unterscheidung von MYH9-bedingten Makrothrombozytopenien (normale Funktion) und von Willebrand Syndromen (abnorme Funktion). Die Diagnose wird durchflusszytometrisch als Fehlen der GPIb/IX-Komplexe auf der Plättchenmembran bzw. molekulargenetisch bestätigt. Besonders tückisch sind die heterozygoten Formen, weil sie wegen der normalen Plättchenfunktion fälschlicherweise als ITP diagnostiziert werden können, wenn nicht gezielter abgeklärt wurde.
Gray platelet Syndrom
Diese wie das BSS autosomal rezessiv vererbte Thrombozytopenie will an dieser Stelle erwähnt sein, um den Stellenwert des Blutausstrichs noch einmal speziell hervorzuheben. Die vergrösserten und in ihrer Anzahl verminderten Plättchen sind vollständig degranuliert, was das charakteristische blassgraue Erscheinungsbild und die entsprechende Namensgebung erklärt. Vorsicht ist hier geboten, dem seltenen Artefakt eines «pseudo-gray platelet syndroms», welches durch Degranulierung der Plättchen wegen prolongierter Wirkung des EDTA im Blutentnahmeröhrchen hervorgerufen wird. Meistens sind aber diese Plättchen nicht derart vergrössert wie im echten Syndrom.
Behandlungsmöglichkeiten
Die Wege zur Optimierung der Hämostase bei hereditären Thrombozytopenien im Sinne eines Konzepts bei Bedarf, reichen von rein prophylaktischen Massnahmen (Verzicht auf Plättchenaggregations-hemmende Medikamente) über supportive und medikamentöse Möglichkeiten (Desmopressin zur Steigerung der Koagulabilität, Tranexamsäure zur Aufhebung der endogenen und lokalen Fibrinolyse).
Copyright bei Aerzteverlag medinfo AG
Klinik für Hämatologie, Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
adrian.bachofner@usz.ch
SYNLAB Suisse SA
Alpenquai 14
6002 Luzern
dimitrios.tsakiris@usb.ch
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Nagler M, Keller P, Siegrist D, Alberio L. A case of EDTA-dependent pseudothrombocytopenia: simple recognition of an underdiagnosed and misleading phenomenon. BMC Clin Pathol 2014;14:19.
2. Noris P, Pecci A. Hereditary thrombocytopenias: a growing list of disorders. Hematology Am Soc Hematol Educ Program 2017(1):385-399
3. Kumar R, Kahr W. Congenital Thrombocytopenia: Clinical Manifestations, Lab- oratory Abnormalities, and Molecular Defects of a Heterogeneous Group of Conditions. Hematol Oncol Clin N Am 2013; 27:465–494
4. Provan D, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia Blood Advances Nov 3;2019(22): 3780-3817
5. Bakchoul T, Marini I. Drug-associated thrombocytopenia. Hematology Am Soc Hematol Educ Program. 2018. Nov 30;2018(1):576-583
6. Neunert C, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Advances Dec 3; 2019 (23): 3829-3866


der informierte @rzt
- Vol. 12
- Ausgabe 5
- Mai 2022