
Veränderungen des roten Blutbildes nach Ausschluss eines Eisen- oder Vitamin B12-Mangels − mit oder ohne Anämie − gehören zu den häufigen diagnostischen Herausforderungen im klinischen Alltag. Aufgrund des sehr breiten Krankheitsspektrums fokussieren wir uns im vorliegenden Beitrag auf Hämoglobinopathien/Thalassämien und myelodysplastische Syndrome.
Veränderungen des roten Blutbildes implizieren sowohl quantitative Abweichungen von den Referenzwerten für die Parameter Hämoglobin (Hb), Hämatokrit (Hk), Erythrozytenzahl (Ec) und -indizes MCV, MCH und MCHC, als auch qualitative Veränderungen der Erythrozytenmorphologie. Von der WHO wurden je nach Alter und Geschlecht Referenzwerte definiert (1).
Mikrozytäre hypochrome Blutbildveränderungen mit oder ohne Anämie
Nach Ausschluss eines Eisenmangels als Ursache einer Mikrozytose und Hypochromie (erniedrigtes MCV und MCH) mit oder ohne Anämie ist differentialdiagnostisch eine Hämoglobinopathie (anomales Hämoglobin oder Thalassämie) zu erwägen (2). Die Inzidenz dieser monogenen Erbkrankheiten wird weltweit mit mehr als 300`000 Geburten pro Jahr geschätzt (3). Betroffen ist vor allem die Bevölkerung aus Ländern mit hoher Malaria-Prävalenz (Tab. 1), was in Folge der Migration aber zunehmend von globaler Relevanz ist. Auch wenn die Mehrheit der Hämoglobinopathien klinisch asymptomatisch ist, ist eine korrekte Diagnose für die betroffenen Patienten wichtig wegen der Vererbung auf Nachkommen bzw. der Familienplanung (vor allem wenn der Partner ebenfalls aus einem Risikogebiet stammt). Besonders essentiell ist die Abgrenzung zu anderen Erkrankungen wie Eisenmangel, der zuerst ausgeschlossen werden sollte, bevor aufwändige Labordiagnostik für Hämoglobinopathien angefordert wird. Eine Eisensubstitution sollte bei Hämoglobinoathien nur bei Nachweis eines Eisenmangels erfolgen, um eine iatrogene Eisenüberladung zu vermeiden.
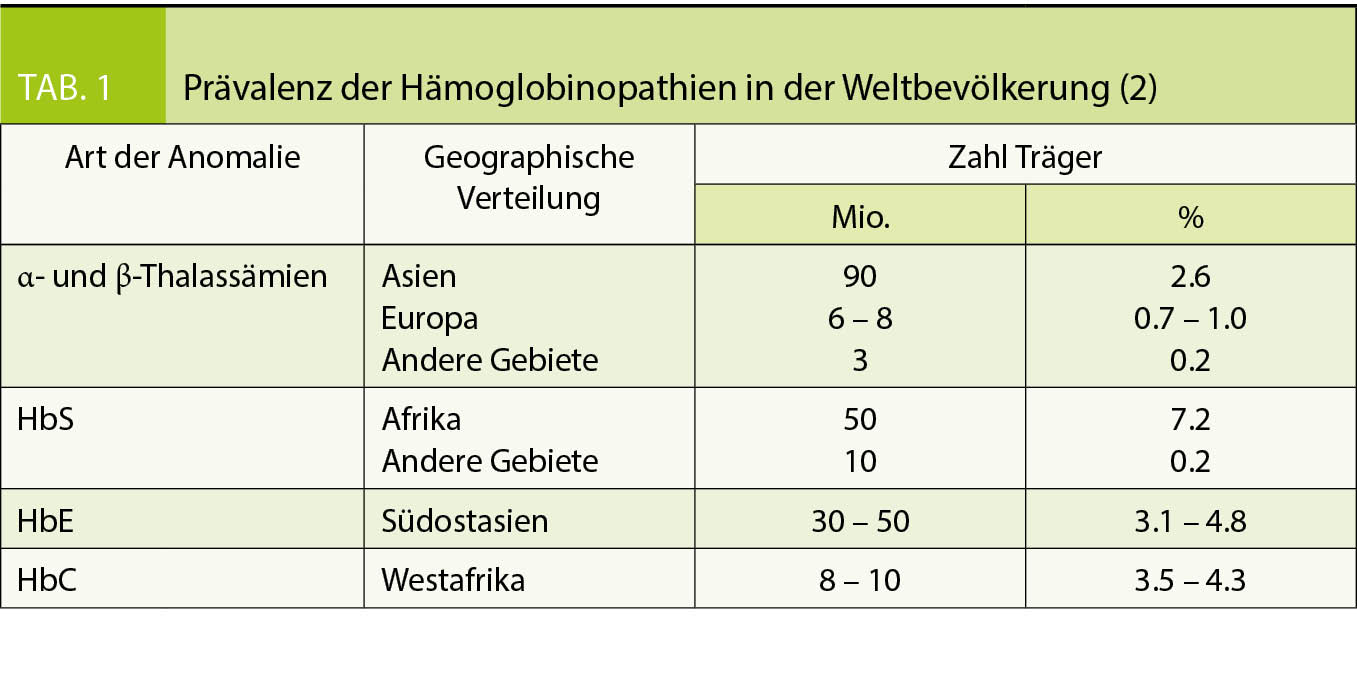
Unterteilt werden die Hämoglobinopathien anhand der zugrundeliegenden molekularen Defekte in anomale Hämoglobine (Hb-Varianten), als Folge einer qualitativen strukturellen Veränderung des Hämoglobins und in Thalassämien als Folge einer quantitativ verminderten Synthese von normalen α- oder β-Globinketten. Es existieren aber Überschneidungen der beiden Gruppen, z.B. thalassämische Hämoglobinopathien wie das HbE.
Die Einteilung der anomalen Hämoglobine erfolgt nach ihren pathophysiologischen Eigenschaften, dem zugrundeliegenden molekularen Defekt, sowie der daraus resultierenden Klinik (Tab. 2). Thalassämien werden nach dem zugrundeliegenden molekularen Defekt in α- und β-Thalassämien unterteilt. Im Blutbild sind Thalassämien mit einer Mikrozytose, Hypochromie und zum Teil einer hyper- bis hyporegenerativen Anämie vergesellschaftet. Die klinische Ausprägung ist je nach zugrundeliegendem Defekt unterschiedlich und entweder besteht eine ineffektive Erythropoiese oder ein vermehrter Abbau der Erythrozyten (Hämolyse). Die Einteilung der β-Thalassämien (minor, intermedia oder major) erfolgt nach Klinik und Restaktivität des β-Gens: β+ mit reduzierter resp. β0 mit fehlender Aktivität des Gens (Tab. 3). Je nach Restaktivität wird kompensatorisch vermehrt HbA2 gebildet und ist diagnosestützend. Die α-Thalassämien werden in die Thalassemia minima, minor, HbH-Krankheit und Hb-Bart’s/Hydrops fetalis eingeteilt. Hier korreliert das Ausmass der Synthesestörung mit der Anzahl nicht-aktiver α-Gene (Tab. 4), wobei eine vollständig fehlende Synthese von α-Ketten nicht mit dem Leben vereinbar ist.
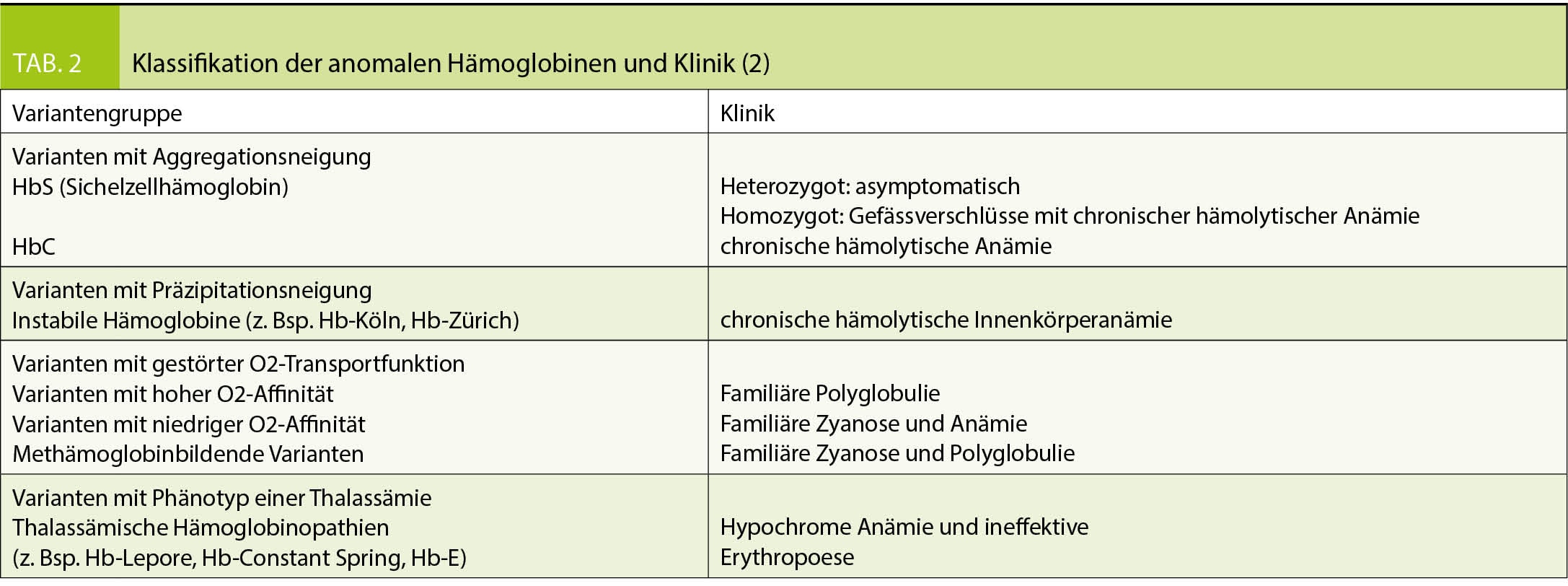
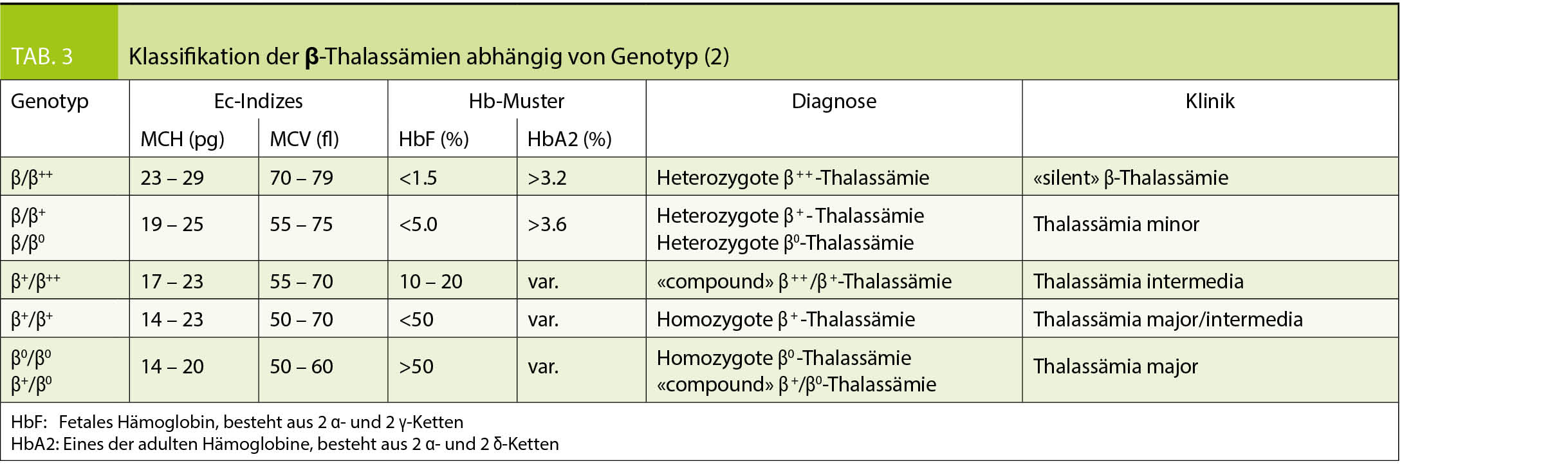
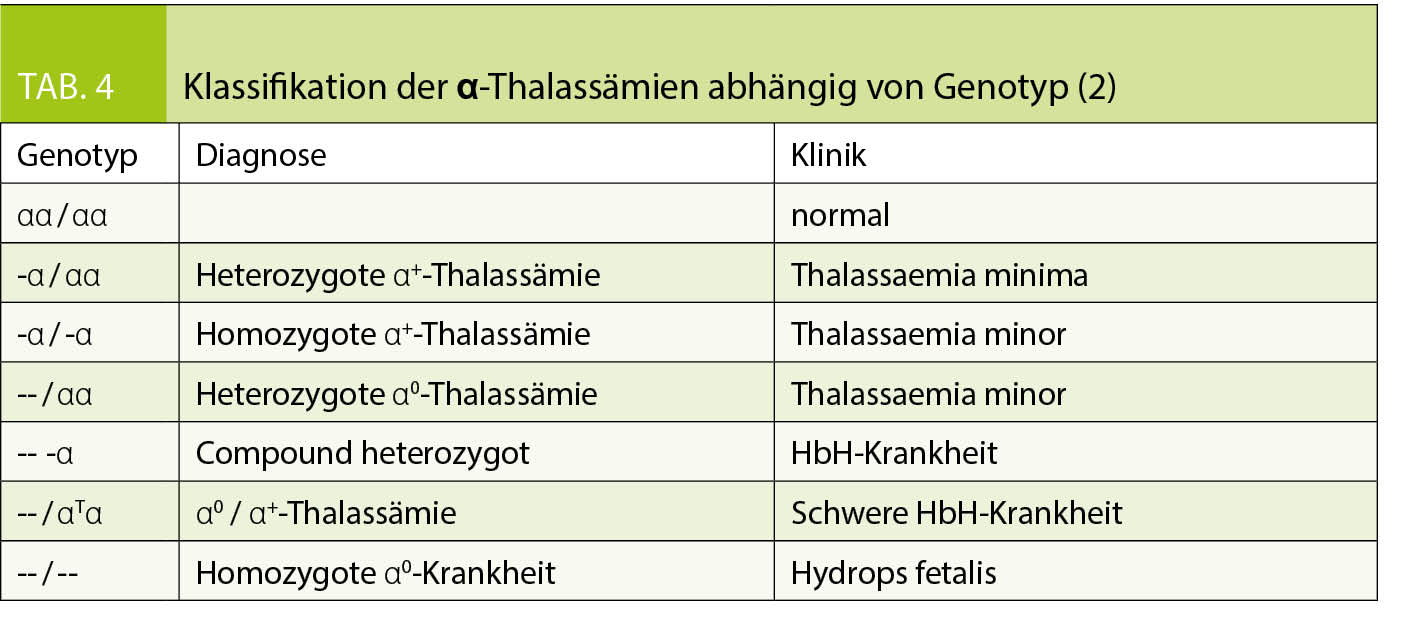
Die Diagnose einer Hämoglobinopathie verlangt die Integration der Klinik (z.B. Gefässverschlüsse, Transfusionsabhängigkeit, Splenomegalie, etc.) und der Laborresultate. Im Labor erfolgt die Abklärung grundsätzlich in 3 Stufen (2, 4, 5):
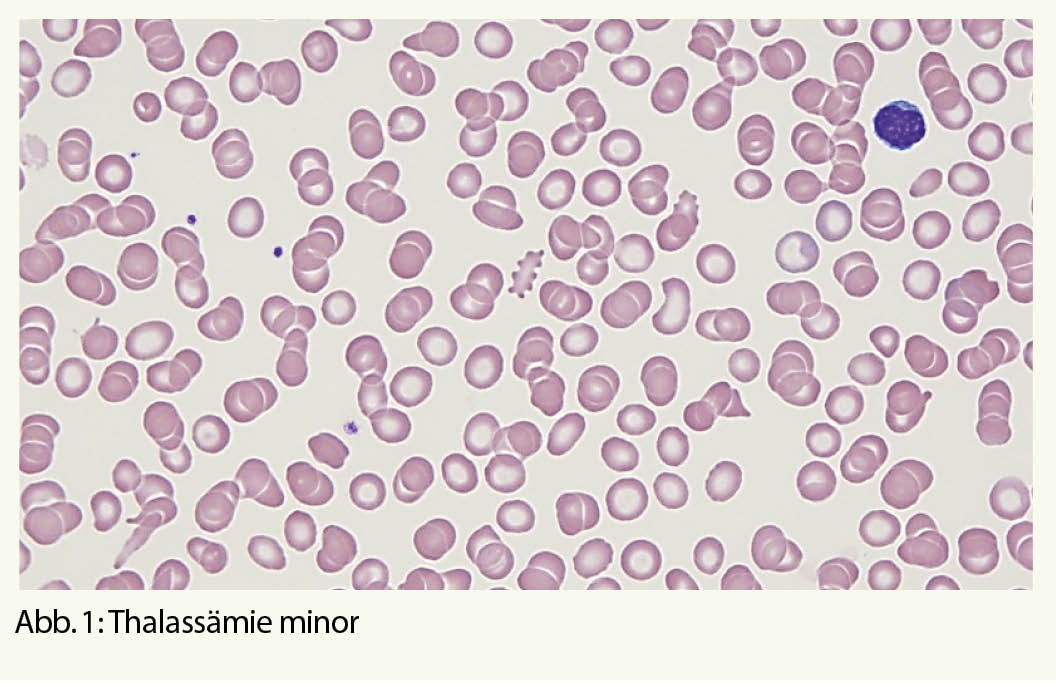
Stufe 1, Basisuntersuchungen: Parameter des roten Blutbildes: Hb, Hk, Ec, MCV, MCH, MCHC, Erythrozyten-Anisozytose (RDW) und Retikulozyten als Marker für die Regeneration der Erythropoiese. Zur Abgrenzung der Haupt-Differentialdiagnose, dem Eisenmangel, Bestimmung der Eisenparameter Ferritin, Transferrin-Sättigung, löslicher Transferrinrezeptor oder Zink-Protoporphyrin. Einige Formeln (Huber-Herklotz/Menzer) weisen eine hohe Sensitivität für das Vorhandensein einer α- resp. β-Thalassämie auf und sind richtungsweisend für die weitere Diagnostik. Ein mikroskopisches Blutbild mit allfälligen Veränderungen wie Mikrozytose und Hypochromie sowie Anisozytose, basophiler Punktierung, Poikilozytose und Polychromasie ist hilfreich (Abb. 1).
Stufe 2, Untersuchungen in einem spezialisierten Labor für Erythrozytenfunktionsdiagnostik: Dazu gehört hauptsächlich die Auftrennung der verschiedenen Hämoglobinfraktionen mittels Chromatographie und Elektrophorese. Die Abgrenzung zwischen HbS und Hb-Varianten, die sich elektrophoretisch und chromatographisch ähnlich verhalten, erfolgt durch den Löslichkeits- und Sicheltest. Weiter ist der Nachweis von HbH-Innenkörpern (überschüssige β-Ketten im Erythrozyten präzipitieren und werden mittels Supravitalfärbung dargestellt) richtungsweisend für eine α-Thalassämie.
Stufe 3, Abklärungen im Labor für medizinische Genetik: Molekularbiologische Analysen ermöglichen eine definitive Diagnose, v.a. bei einer α –Thalassämie spezifizieren sie die Diagnose einer Hämoglobinopathie. Untersucht wird ein zugrundeliegender genetischer Defekt auf den Chromosomen 11 (für β-Thalassämien) und 16 (für α-Thalassämien).
Eine erworbene Thalassämie kommt selten vor, und zwar beim myelodysplastischen Syndrom (MDS) mit Mutation des ATRX-Gens (Xq21.1). Dabei wird die Expression des α-Globingens herunterreguliert (5).
Makrozytäre Blutbildveränderungen mit oder ohne Anämie
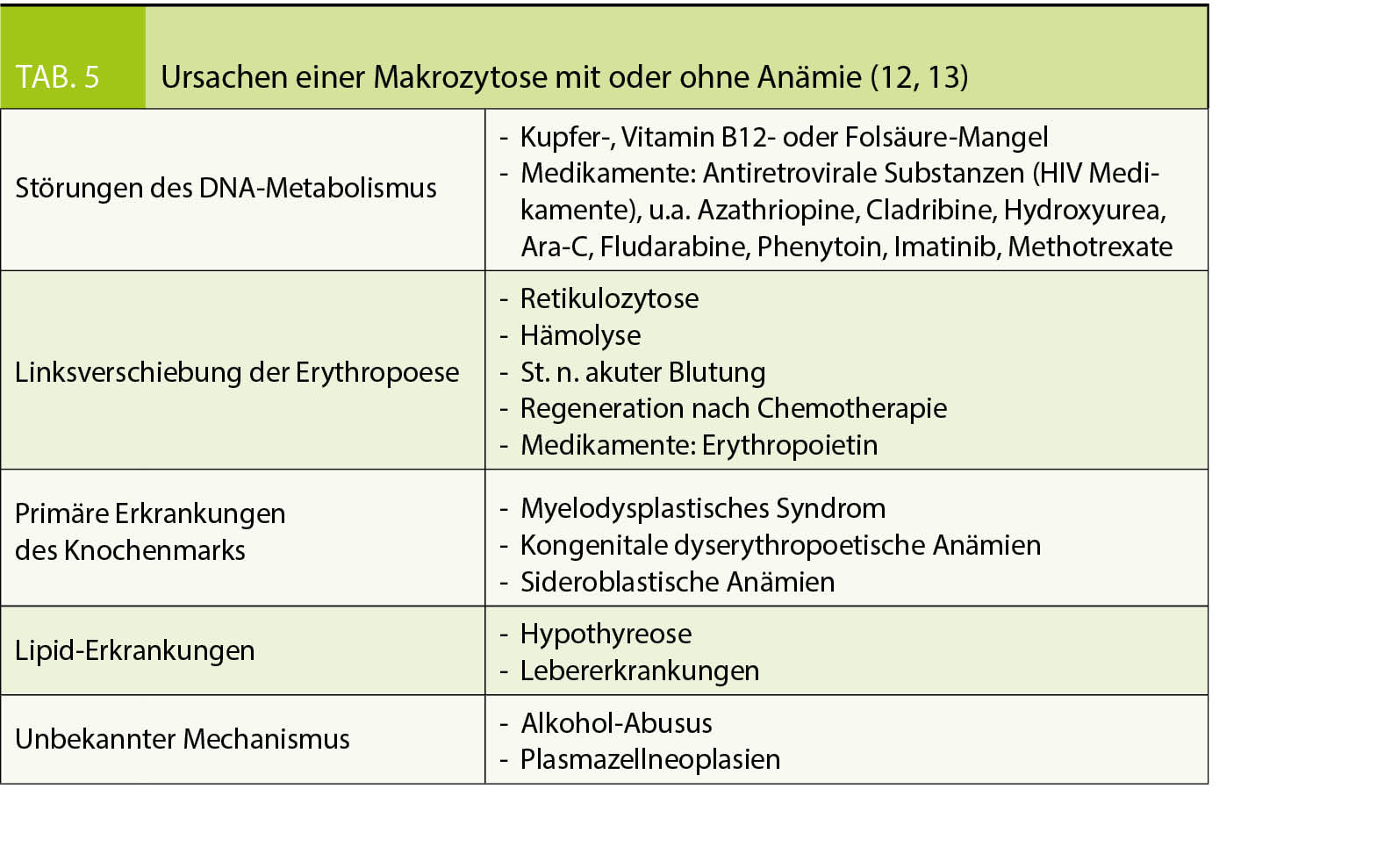
Zugrundeliegende Ursachen, die zu einer Markozytose führen, sind vielfältig und deren pathophysiologischen Mechanismen z.T. nicht vollständig verstanden (Tab. 5):
- DNA-Synthesestörung, entweder bedingt durch einen Mangel an Substraten oder durch die Einnahme von bestimmten Medikamenten
- Gesteigerte Erythropoiese mit Retikulozytose (Retikulozyten sind grösser als reife Erythrozyten)
- Hereditäre (z.B. hereditäre Stomatozytose) oder erworbene (im Rahmen einer Lebererkrankung) Erythrozytenmembranerkrankungen
- Primäre Erkrankungen des Knochenmarks, z. B. MDS.
Bleibt die Ätiologie der Makrozytose nach der Medikamenten-anamnese und nach Ausschluss eines Substratmangels oder einer bekannten Hämolyse / Retikulozytose unklar, ist zur erweiterten Diagnostik oft eine fachärztliche hämatologische Untersuchung sinnvoll.
Mit zunehmendem Alter steigt die Wahrscheinlichkeit eines MDS mit einer Inzidenz von 0.5/100 000 bei Patienten < 50 Jahren und 89/100 000 bei Patienten > 80 Jahren (6). Männer sind etwas häufiger betroffen als Frauen mit Ausnahme des MDS mit isolierter Deletion 5q. Gekennzeichnet ist ein MDS durch eine ineffektive und dysplastische Hämopoiese als Folge von klonalen Veränderungen der hämatopoetischen Stammzellen im Knochenmark.
Klinisch präsentieren sich die Patienten teils mit Allgemeinsymptomen und / oder mit Beschwerden, die mit vorliegenden Zytopenien assoziiert sind, bei einer Anämie z.B. Fatigue, Anstrengungsdyspnoe und Angina pectoris.
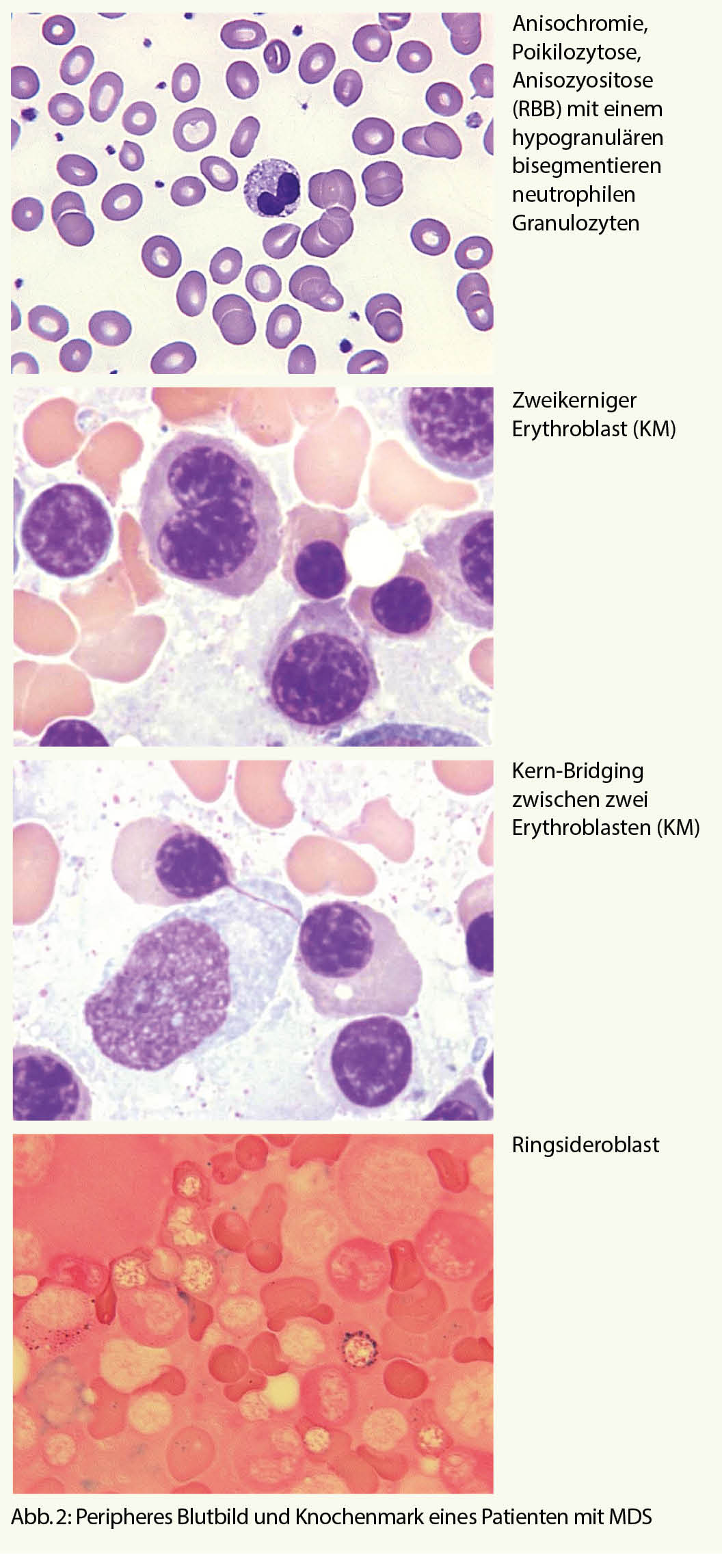
Im Blutbild zeigt sich oft eine progrediente, meist makrozytäre, hyporegenerative (d.h. mit inadäquat tiefer Retikulozytenzahl) Anämie. Mikroskopisch sind die Erythrozyten makrozytär mit Anisozytose, Anisochromie, Poykilozytose (Abb. 2). Blasten können vorhanden sein, jedoch darf der Wert 19% nicht überschreiten, ansonsten liegt eine akute myeloische Leukämie vor.
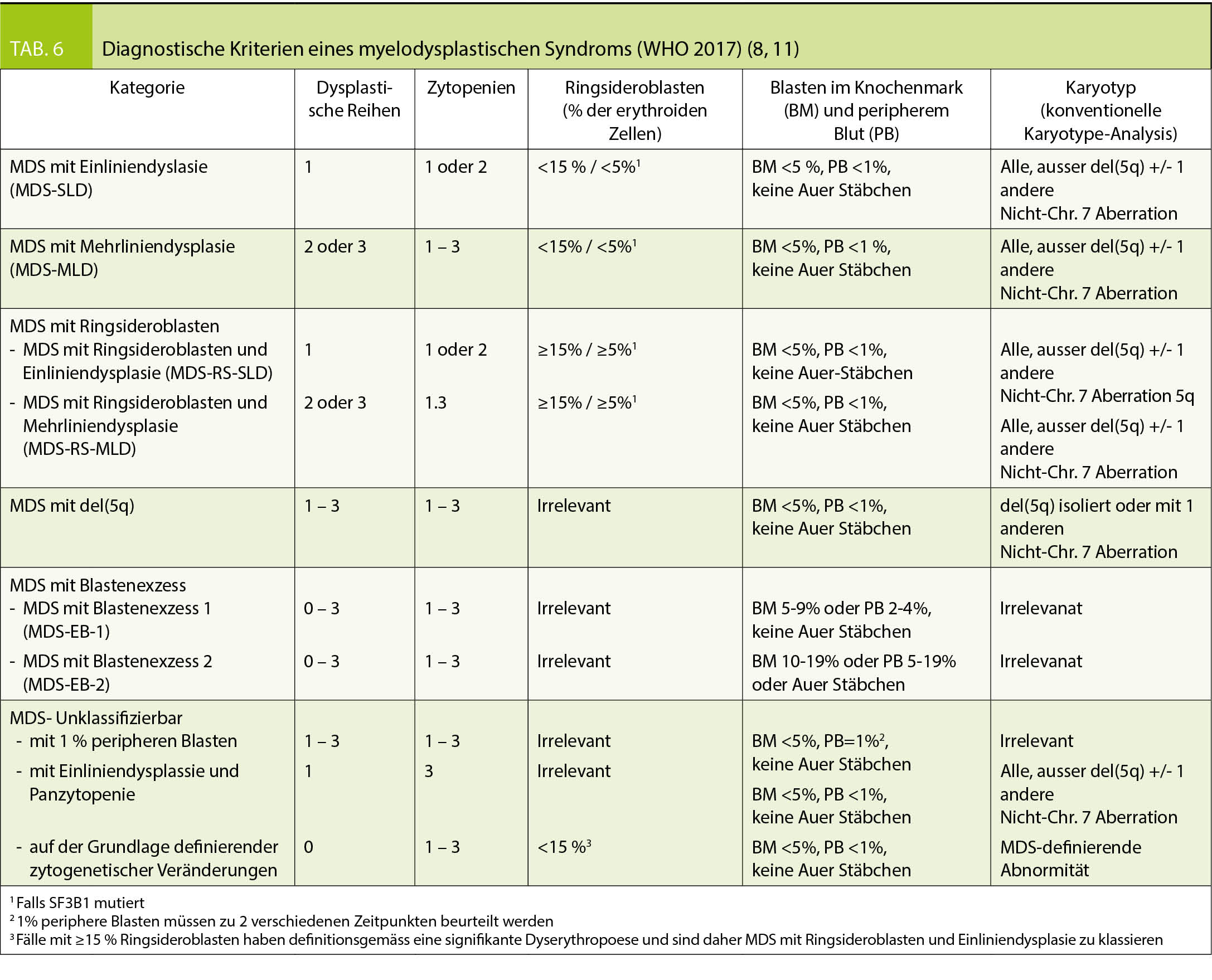
Um die Verdachtsdiagnose MDS bestätigen oder ausschliessen zu können ist eine Untersuchung des Knochenmarks nötig. Gemäss Klassifizierung der WHO 2017 (8) werden für die Einteilung der MDS die Prozentzahl der Blasten im peripheren Blut und im Knochenmark, der Dysplasiegrad der drei Zellreihen im Knochenmark, sowie genetische Befunde benötigt (Tab. 6). Folgende Veränderungen gelten als Kriterium für Dysplasie der Erythropoiese:
- Veränderungen der Kernmorphologie: Knospung, internukleäres Bridging, mehrkernige Formen, Kernpyknose, megaloblastäre Veränderungen (Abb. 2)
- Veränderungen des Zytoplasmas: Vakuolisierung, Ringsideroblasten (Abb. 2)
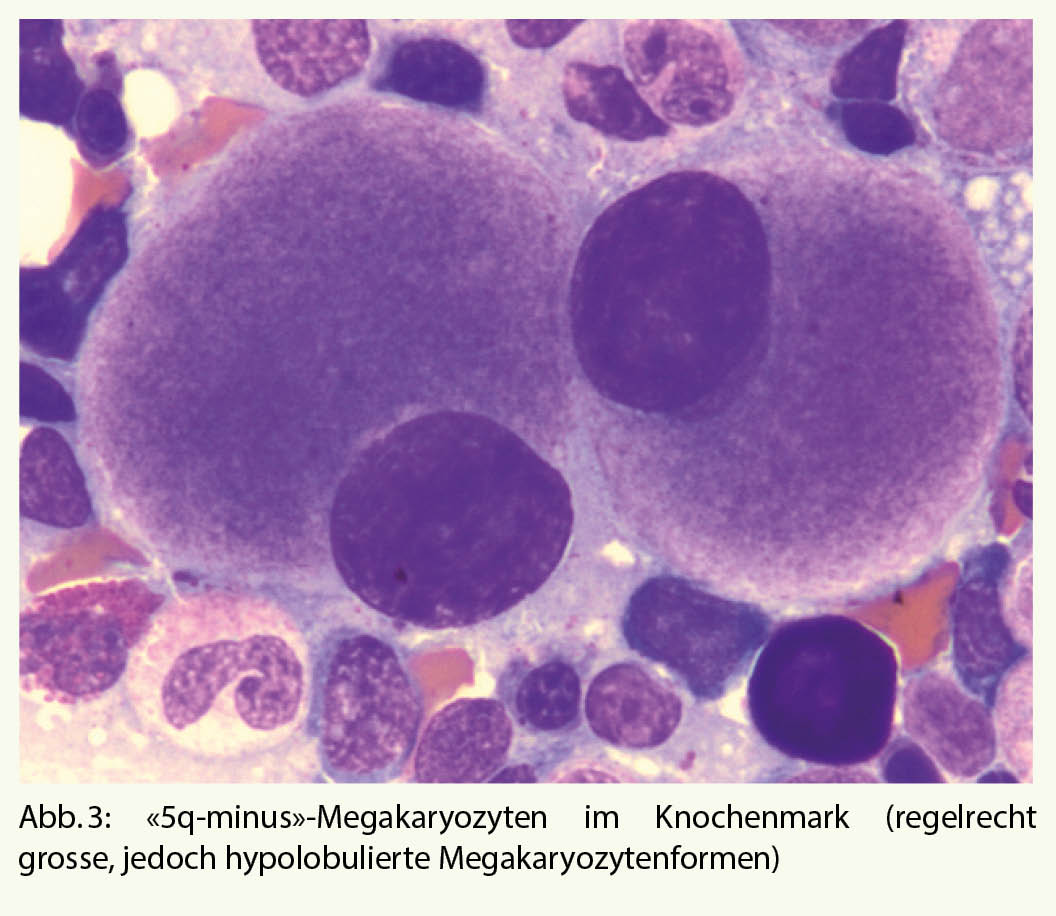
Dann stellt sich die Frage, ob die dysplastischen Anzeichen durch eine klonale Veränderung (vs. medikamentös, toxisch) bedingt sind. Bei ca. 90% der MDS-Fälle wird eine klonale zytogenetische Veränderung festgestellt (7). Die zytogenetischen und molekularbiologischen Befunde aus dem Knochenmark sind aber nicht nur für die definitive Diagnose, sondern zum Teil auch für die Prognose und die Wahl der Therapie des MDS entscheidend. Als Beispiel dient das MDS mit isolierter Deletion des langen Armes vom Chromosom 5 (sogenanntes «5q-minus-Syndrom»): Es zeigen sich charakteristische Veränderungen des Blutbildes mit normo- bis makrozytärer Anämie und normaler bis erhöhter Thrombozytenzahl, im Knochenmark auffällige, regelrecht grosse, einkernige Megakaryozyten (Abb. 3). Die Prognose ist günstig, und Behandlung der Wahl ist eine immunmodulatorische Therapie (Lenalidomid).
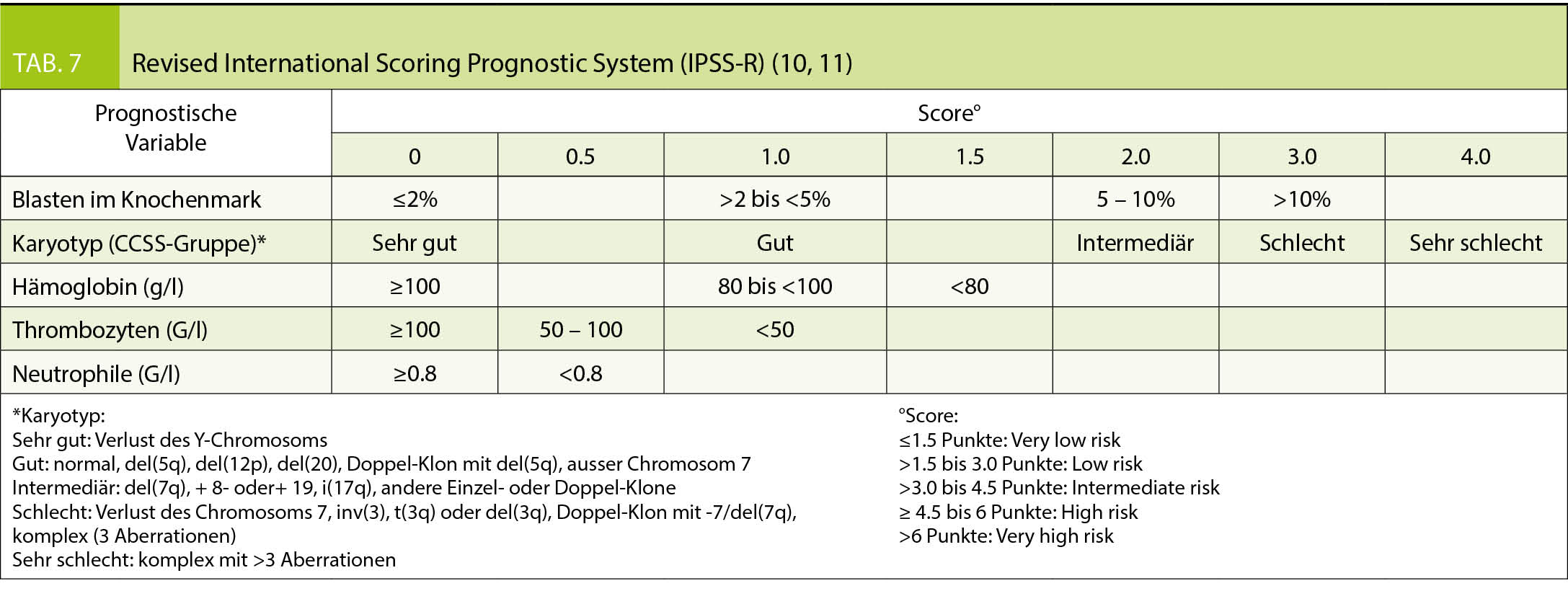
Bei MDS Patienten erlaubt der revidierte IPSS Score (Tab. 7) durch Integration morphologischer und zytogenetischer Befunde eine Prognose des Krankheitsverlaufs (9, 10).
Die lange Zeit eher beschränkten therapeutischen Optionen bei MDS (in kurativer Absicht eine allogene Stammzelltransplantation vs. palliative / supportive Massnahmen wie Transfusionen oder Wachstumsfaktoren) haben sich in den letzten 20 Jahren verbessert. Zu nennen sind insbesondere die Eisenchelation sowie neue immunmodulatorische, hypomethylierende oder gar auf ein molekulares Target gezielt wirkende Medikamente. Dabei handelt es sich um relativ gut verträgliche, ambulant durchführbare mehrheitlich orale Therapien, die durchaus auch alten und komorbiden Patienten eine verbesserte Lebensqualität oder Lebensverlängerung bieten können.
Copyright bei Aerzteverlag medinfo AG
Institut für Labormedizin
Kantonsspital Aarau
Tellstrasse 25
5001 Aarau
adriana.mendez@ksa.ch
Institut für Labormedizin
Kantonsspital Aarau
Tellstrasse 25
5001 Aarau
Institut für Labormedizin
Kantonsspital Aarau
Tellstrasse 25
5001 Aarau
Die Autorinnen haben im Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
1. Blanc B, Finch CA, Hallberg L, et.al. Nutritional anaemias. Report of a WHO Scientific Group. WHO Tech Rep Ser. 1968; 405: 1-40.
2. Huber AR, Ottiger C, Risch L, et.al. Hämoglobinopathien: Pathophysiologie und Klassifizierung. Schweiz Med Forum 2004: 4:895-901
3. Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331-4336.
4. Huber AR, Ottiger C, Risch L, et.al. Thalassämie-Syndrome: Klinik und Diagnose. Schweiz Med Forum 2004: 4:947-952
5. Mullen S, Talaulikar D. Acquired α-thalassemia associated with myelodysplastic syndromes. Blood. 2018;132(20):2209.
6. Williamson PJ, Kruger AR, Reynolds PJ, Hamblin TJ, Oscier DG. Establishing the incidence of myelodysplastic syndrome. Br J Haematol. 1994;87(4):743-745.
7. Will B, Zhou L, Vogler TO, et al. Stem and progenitor cells in myelodysplastic syndromes show aberrant stage-specific expansion and harbor genetic and epigenetic alterations. Blood. 2012;120(10):2076-2086.
8. Swerdlow S, et.al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th Edition. 2017
9. Greenberg P, Cox C, LeBeau MM, et.al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997 Mar 15;89(6):2079-88.
10. Greenberg PL, Tuechler H, Schanz J, et.al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012 Sep 20;120(12):2454-65.
11. https://www.onkopedia.com/de/onkopedia/guidelines/myelodysplastische-syndrome-mds/@@guideline/html/index.html
12. Hoffbrand V, Provan D. ABC of clinical haematology. Macrocytic anaemias. BMJ. 1997;314(7078):430-433.
13. d’Onofrio G, Chirillo R, Zini G, et.al.. Simultaneous measurement of reticulocyte and red blood cell indices in healthy subjects and patients with microcytic and macrocytic anemia. Blood. 1995;85(3):818-823.



der informierte @rzt
- Vol. 10
- Ausgabe 9
- September 2020