Über 6200 Mamma- und Ovarialkarzinome werden jährlich in der Schweiz diagnostiziert (1). Ca. 5-10% der diagnostizierten Mammakarzinome und 10-15% der epithelioiden Ovarialkarzinome haben eine genetische Ursache (2). Die Grundlage einer genetischen Ursache aller Krebsarten liegt an Varianten in bestimmten Genen, die meistens an der Regulierung des Zellwachstums oder der DNA-Reparatur beteiligt sind (3). Nicht alle dieser Varianten sind von einem Elternteil geerbt. Es können auch neue Varianten zum ersten Mal in einer Keimzelle oder im befruchteten Ei selbst während der frühen Embryogenese auftreten Zudem können sporadische Varianten auch erst in (somatischen) Tumorzellen auftreten (4).
Over 6200 mammary and ovarian carcinomas are diagnosed annually in Switzerland (1). Approximately 5-10% of diagnosed breast carcinomas and 10-15% of epithelioid ovarian carcinomas have a genetic cause (2). The basis of a genetic cause of all cancers is due to variants in certain genes, most of which are involved in the regulation of cell growth or DNA repair (3). Not all of these variants are inherited from a parent. New variants may also appear for the first time in a germ cell or in the fertilized egg itself during early embryogenesis In addition, sporadic variants may also first appear in (somatic) tumor cells (4).
Key Words: Genetic counseling, Genetics, breast cancer, ovarian cancer, germline mutation
Familienstudien haben seit langem bewiesen, dass die Verwandten ersten Grades (d.h. Eltern, Geschwister und Kinder) und zweiten Grades (d.h. Grosseltern, Tanten oder Onkel, Enkel, Nichten oder Neffen) der von Tumorleiden betroffenen Personen ein höheres Risiko einer Tumorerkrankung haben. Diese Individuen können eine erhöhte Anfälligkeit für Krebs infolge einer oder mehrerer Genvarianten erben, welche in den elterlichen Keimbahnzellen vorhanden sind. Krebs, der sich bei diesen Personen entwickelt, kann als erblicher oder familiärer Krebs klassifiziert werden.
In diesem Bereich spielt eine genetische Beratung eine wichtige Rolle, das Risiko eines erblich bedingten Tumors zu erkennen. Ziel dafür ist, rechtzeitig mögliche Vorbeugungsmassnahmen unternehmen zu können.
Die genetische Beratung – Grundlagen
Mamma- und Ovarialkarzinome sind zwei gynäkologische Tumorerkrankungen, bei denen wir es häufig mit einem erblichen Hintergrund zu tun haben. Bei Patientinnen ohne Genvarianten beträgt das Lebenszeitrisiko eines Mammakarzinoms etwa 12-13%, während das Risiko eines Ovarialkarzinoms bei 1-2% liegt. Bei Patientinnen mit bestimmten Genvarianten ist das Risiko beider Tumoren deutlich erhöht. Unter HBOC (Hereditary Breast and Ovarian Cancer Syndrome) versteht man eine klinische Bedingung, bei der sowohl Brust- als auch Ovarialkarzinome deutlich häufiger als in der Allgemeinpopulation auftreten. Dies liegt an bestimmten Genmutationen, welche in der Familie vererbt werden können. Das führt dazu, dass diese Tumorleiden meist auch mehrere Familienmitglieder betreffen.
Unter Berücksichtigung bereits aufgetretener Erkrankungsfälle in der Familie und spezieller Zusatzkriterien (z.B. Erkrankungsalter, Bilateralität oder Histologie des Tumors), kann eine Wahrscheinlichkeit für eine vererbte Tumorerkrankung wie HBOC für die betreffende Person ermittelt werden.
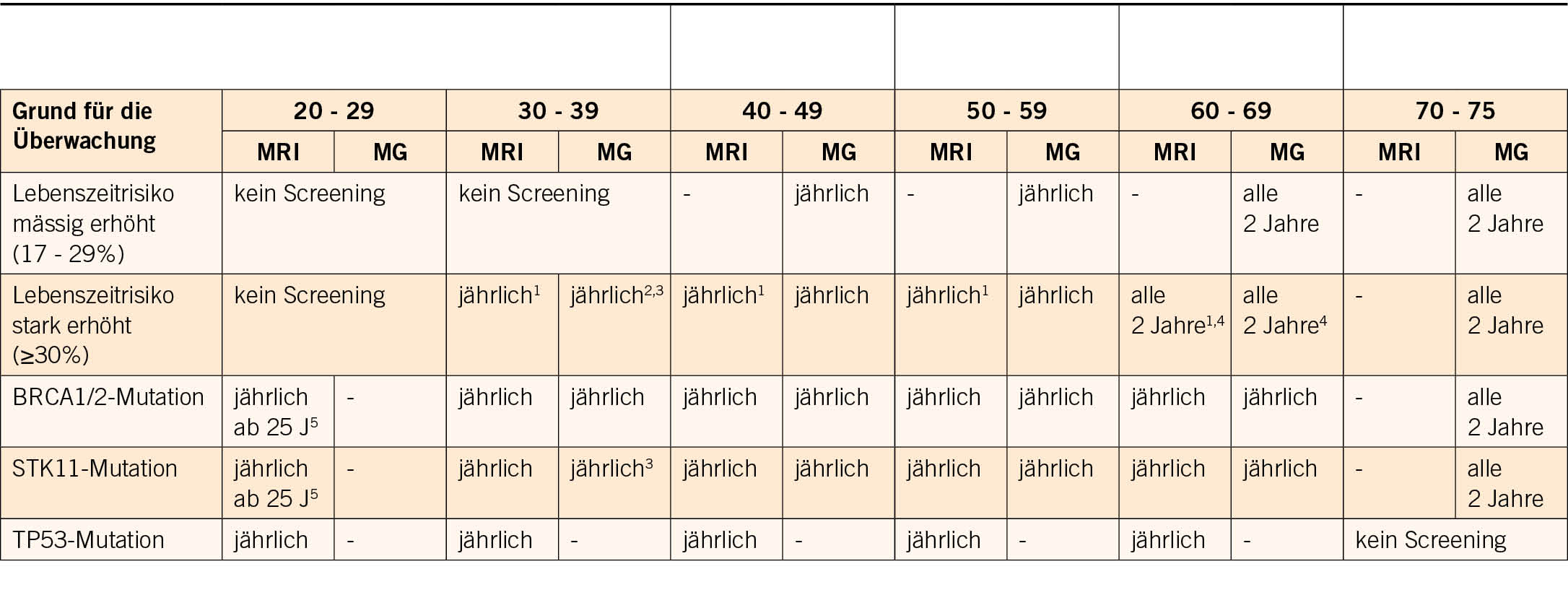
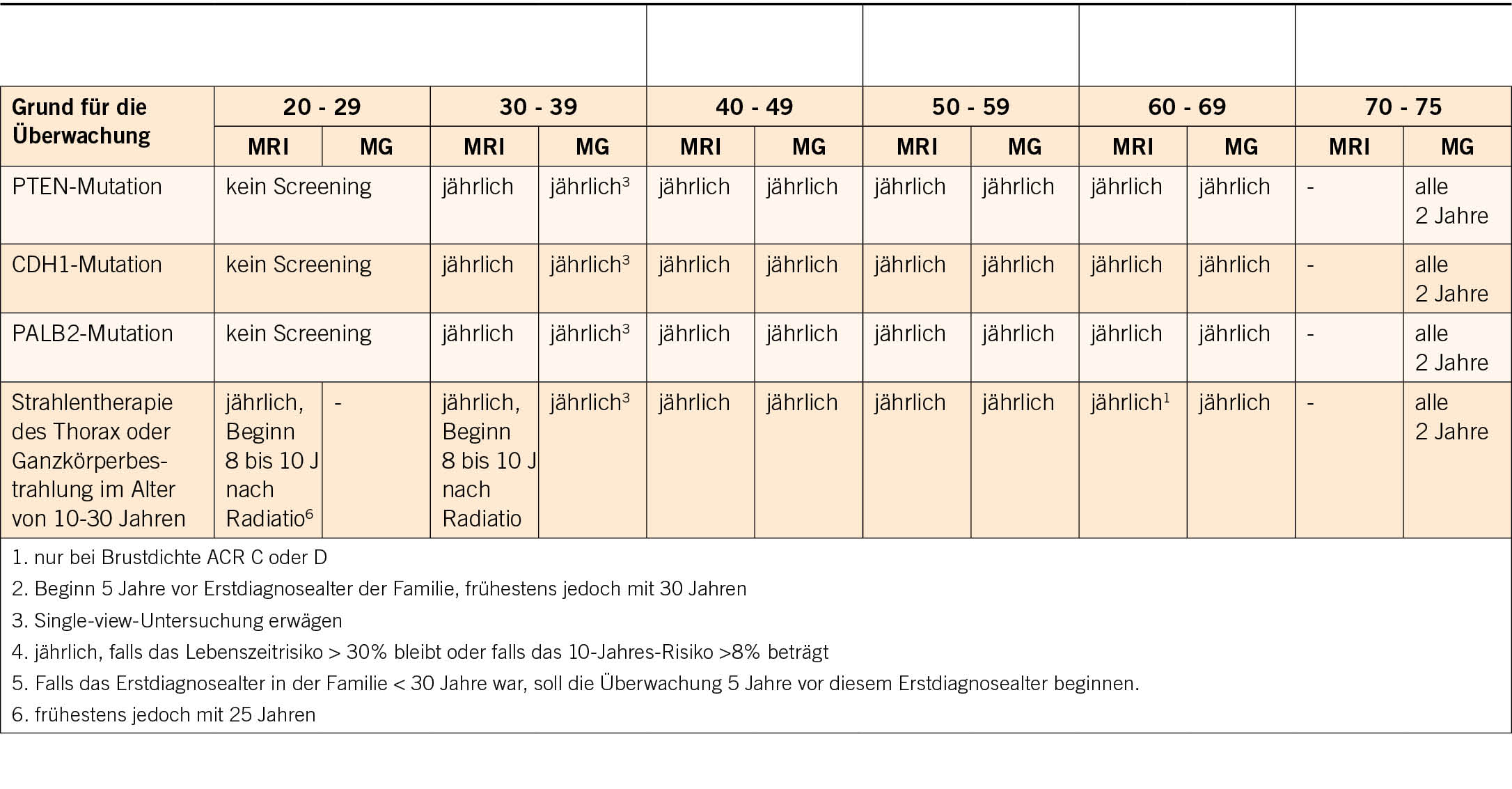
Bei der genetischen Beratung wird das Risiko geschätzt, und je nach Höhe des Risikos wird bei der Krankenkasse eine Kostengutsprache für eine genetische Ersttestung beantragt. Im ersten Schritt wird mit der Patientin die persönliche Anamnese (Risiko- und Schutzfaktoren für/von Tumoren, benigne Erkrankungen/Zeichen mit Hinweis auf einen genetischen Hintergrund) und der Familienstammbaum erhoben. Es werden auch ausführlich die Bedeutung dieser Testung und die möglichen Implikationen diskutiert. In den neu adaptierten und erweiterten Schweizer Richtlinien (4) werden die Bedingungen auf der Basis von Stammbaum und Tumorkriterien aufgelistet, wann eine genetische Testung zulasten der Krankenkasse indiziert ist (Tab. 1).
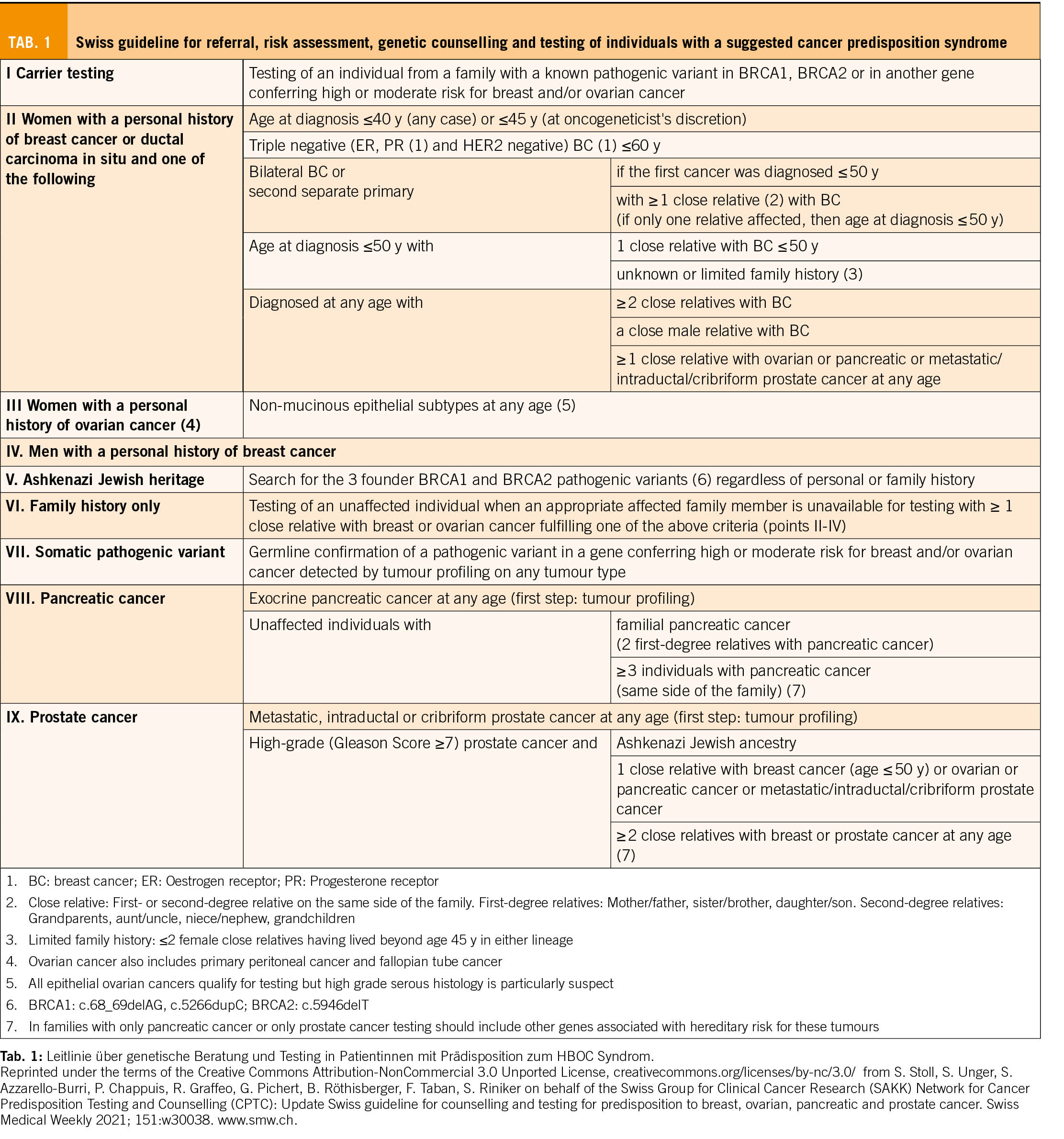
Zusätzlich zur Beachtung dieser Indikationsliste werden stammbaumbasierte Risikoberechnungen mit verschiedenen Computerprogrammen durchgeführt (z.B. werden in Basel CancerGeneTM und CanRiskTM verwendet). Das damit errechnete Risiko, entspricht der Wahrscheinlichkeit, dass Mutationen in bestimmten Genen zu finden sind. Darüber hinaus kann mit diesen Softwareprogrammen auch das Risiko einer Tumorerkrankung in 5-10 Jahren und das Lebenszeitrisiko für bestimmte Tumoren berechnet werden, was speziell auch in der Beratung von Ratsuchenden hilft, bei denen schlussendlich keine genetische Veranlagung gefunden wird. Wenn das Risiko für das Vorliegen einer Mutation in diesen Genen >5-10% ist, ist die Indikation für eine genetische Testung auch gegeben.
Da sich die Kosten für eine genetische Ersttestung auf ca. 4000.- sFr. belaufen, wird, wenn immer möglich, vorgängig eine Kostengutsprache bei der Krankenkasse dafür eingeholt. Der Test selbst erfolgt durch eine Blutentnahme (2 Röhrchen EDTA-Blut), die Analyse nimmt aber mehrere Wochen in Anspruch, weshalb bei Tumorpatientinnen möglichst früh an eine genetische Testung gedacht werden soll, damit das Resultat insbesondere für die operativen Entscheide rechtzeitig vorliegt.
Für eine genetische Keimbahntestung muss ein schriftliches informiertes Einverständnis der betroffenen Person auf jeden Fall vorliegen. Es kann deshalb in Situationen mit fortgeschrittenen Tumorerkrankungen auch einmal sinnvoll sein, das Einverständnis einer Patientin für eine DNA-Asservierung für eine spätere (posthum) von den Nachkommen gewünschte genetische Untersuchung einzuholen. Genetische Beratungen bei Tumorpatientinnen können von allen mit dem Krankheitsbild vertrauten Ärzte/Ärztinnen angeboten werden unter Berücksichtigung/Erfüllung der gesetzlich verlangten Dokumentation der entsprechend notwendigen Inhalte. Genetische Beratungen bei Gesunden (für prädiktive Testungen zu Lasten der Krankenkassen) sind Ärzten/ Ärztinnen mit einem Facharzttitel in Medizinischer Genetik oder Mitgliedern des Netzwerkes für genetische Testung auf Krebsprädisposition und Risikoberatung (CPTC) der Schweizerischen Arbeitsgemeinschaft für Klinische Krebsforschung (SAKK) vorbehalten (www.sakk.ch).
Während Medizinische Genetiker/Genetikerinnen (SGMG) sich auf die Beratung sämtlicher erblich bedingter Krankheiten spezialisiert haben, beraten die Mitglieder des SAKK CPTC Netzwerkes nur Patientinnen/Patienten und deren Familien, bei denen es um erbliche Krebserkrankungen geht (4).
2017 wurden erstmals Schweizer Richtlinien über die Beratung und Testung bei genetisch bedingtem Brust- und Eierstockkrebs veröffentlicht und 2021 von der CPTC der SAKK aktualisiert (Tab. 1).
Folgende Aspekte sind von entscheidender Bedeutung:
- frühes Alter des Auftretens von Krebs
- erhöhte Zahl von Krebsfällen über Generationen hinweg
- Bilateralität der Tumorerkrankung
- Auftreten mehrerer typischer Tumoren bei derselben Person oder nahen Verwandten
- besondere ethnische Herkunft (z.B. Ashkenazim-jüdische Abstammung)
- Histologie
Folgende Themen sind bei der Beratung zu erläutern:
- Aufklärung des Vererbungsmusters
- verfügbare Testmöglichkeiten
- mögliche Befunde (pathogene Varianten, Varianten mit
- unklarer Bedeutung VUS)
- Krankheitsmanagement
- gezielte Behandlung
- Überwachungs- und Präventionsmöglichkeiten.
Die genetische Testung – das Vorgehen
In den letzten Jahren wurden mehrere Gene identifiziert, welche mit erblicher Anfälligkeit für Brust- und Eierstocktumoren assoziiert sind. Die Hochrisikogene, deren Mutationen am häufigsten solche Tumoren verursachen sind BRCA1, BRCA2 (Brust und Ovar) und PALB2 (Brust). Diese werden häufig zuerst auf mögliche Veränderungen getestet, da die empfohlenen Konsequenzen bei Vorliegen einer Mutation für die Primärtherapie eine Rolle spielen und auch schon mit Studien belegt sind und nur noch wenig unklare Varianten gefunden werden (3-5%).
Von besonderem Interesse ist die Tatsache, dass diese Genveränderungen nicht nur mit gynäkologischen Tumoren korrelieren, sondern auch mit anderen Malignomen wie männlichem Brustkrebs, Prostatakarzinom und Pankreaskarzinom (5).
Wenn bei einem zweizeitigen Vorgehen in den Genen BRCA1, BRCA2 oder PALB2 keine pathogene Mutation gefunden wird, kann/soll je nach Ausgangssituation eine Erweiterung auf ein Panel von weiteren selterenen Hochrisiko- oder Mittelrisiko-Genen erweitert werden (z.B. unser Panel in Basel: ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, MLH1, MSH2, MSH6, PALB2, PTEN, RAD51C, RAD51D, STK11 und TP53). Diese Veranlagungen können seltene Krebssyndrome verursachen wie z.B. Li-Fraumeni-, Cowden-, Peutz-Jeghers- oder auch Lynch-Syndrome. Diese sind nicht nur mit Brust- bzw. Ovarialkrebs verbunden, sondern auch mit anderen Tumoren wie Melanomen, Kolonkarzinomen und Prostatakarzinomen.
Die Auswahl der Gene, die in einer Testung berücksichtigt werden, soll auf den persönlichen und familiären Merkmalen basieren, welche die Wahrscheinlichkeit eines Tumorleidens vorhersagen. In der Beratung ist es häufig so, dass in der Familie einer Ratsuchenden noch keine pathogene Genvariante bekannt ist. Da ist es am besten, wenn zunächst ein von einem Tumor betroffenes Familienmitglied getestet wird, da bei dieser Person die höchste Wahrscheinlichkeit besteht, eine vorhandene familiäre Veranlagung auch zu finden.
Heute werden mehrere Gene mit Next-Generation-Sequencing-Technologien (NGS) gleichzeitig getestet. Ein Multi-Gen-Panel-Test ist effizienter und kostengünstiger in der Bestimmung von potentiell oder klar pathogenen Genvarianten.
Nicht alle Genevarianten in einem Risikogen haben aber eine eindeutige Korrelation zu einem erhöhten Krebsrisiko. Auch in den bekannten mit Tumoren assoziierten Genen gibt es Varianten unklarer Signifikanz (VUS), die zwar nicht dem Bevölkerungsdurchschnitt entsprechen, aber lediglich gutartige Polymorphismen sind, welche nicht mit einem erhöhten Krebsrisiko einhergehen. Deshalb werden bei unklaren Varianten primär KEINE Massnahmen abgeleitet und sie sollen NICHT in Therapieentscheide einbezogen werden. Es macht aber Sinn, nach einigen Jahren nochmals die genetischen Datenbanken abfragen zu lassen, was bis dahin über die unklare Variante bekannt ist, so dass diese möglicherweise neu klassiert und die Empfehlungen angepasst werden können.
Nach der Testung: mögliche Massnahmen
Mutationen in BRCA1, BRCA2 und PALB2
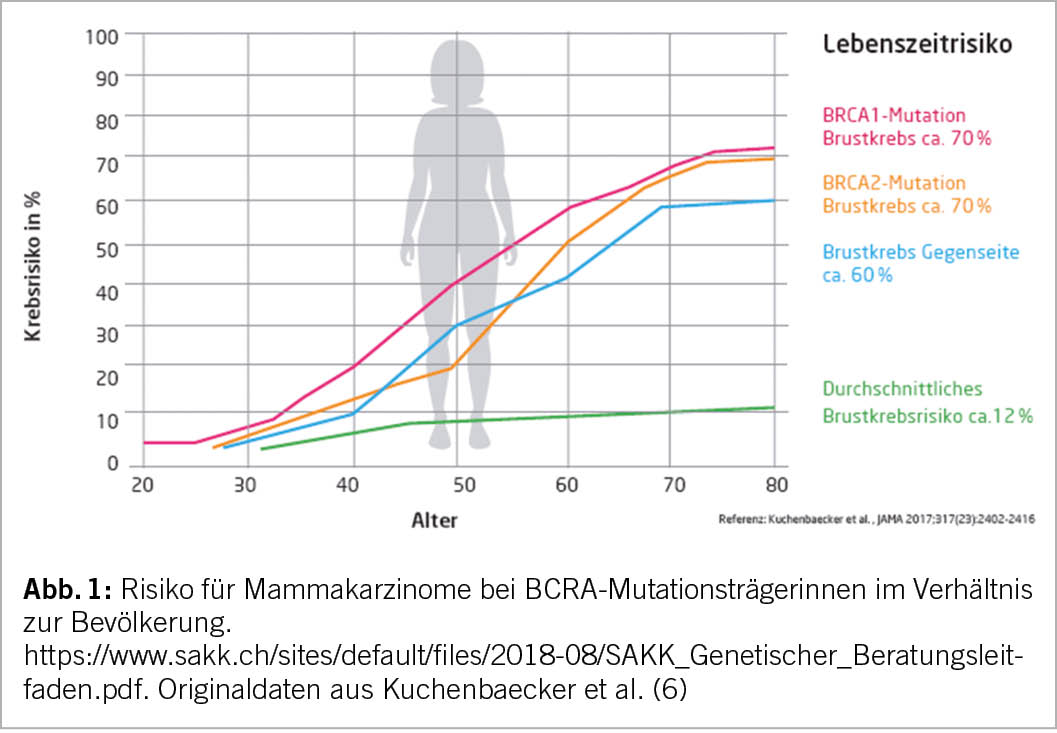
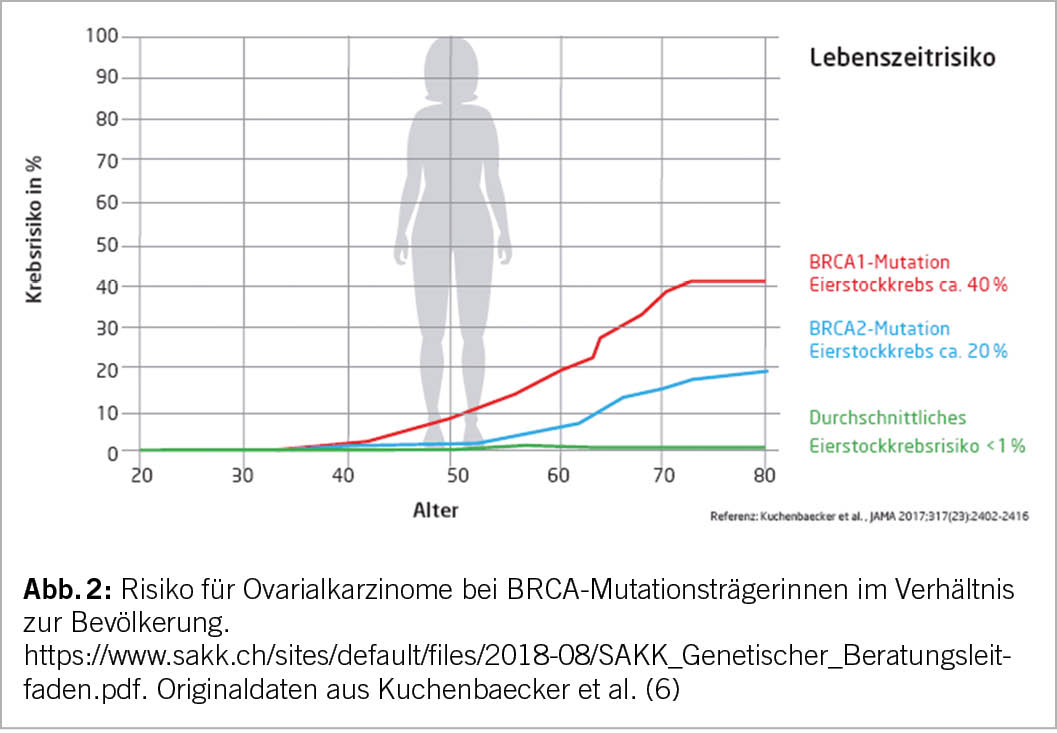
Ein hohes Lebenszeitrisiko einer Tumorerkrankung wird als >30% für Mammakarzinome und 3-5% für Ovarialkarzinome definiert. Eine pathogene Mutation in BRCA1, BRCA2 und PALB2 korreliert in der Regel mit einem hohen Lebenszeitrisiko für Tumoren.
Nach Erkennen eines stark erhöhten Krebsrisikos bei BRCA1– oder BRCA2-Mutationsträgerinnen können je nach Lebenssituation prophylaktische operative Eingriffe im Sinne einer bilateralen Mastektomie und/oder einer bilateralen Salpingoophorektomie (Adnexektomie) angeboten werden mit einer Risikoreduktion für die entsprechenden Tumoren von über 95% (als kassenpflichtige Leistung).
Eine Studie hat vor kurzem bewiesen, dass eine bilaterale Mastektomie speziell in jungen Patientinnen mit pathogener BRCA-1-Genvariante zu einer reduzierten Sterblichkeitsrate im Vergleich zu einer einfachen Früherkennung mit seriellen Kontrollen führt (7).
Alternativ stehen hinsichtlich des Mammakarzinoms (leider nicht für Ovarialkarzinome) Früherkennungsprogramme zur Verfügung mit der Idee, Tumoren zwar nicht verhindern, aber in einem günstigeren Stadium erkennen zu können (4,8). Als weitere Option kann das Risiko eines Mamakarzinoms durch eine 5-jährige Chemoprävention mit einem Antihormon (i.d.R. Tamoxifen) um 30% reduziert werden.
Bei pathogenen PALB2-Mutationsträgerinnen ist das Lebenszeit-Risiko für ein Mammakarzinom auch deutlich erhöht (ca. 50%), aber nicht so hoch wie bei Mutationen in BRCA1 oder BRCA2. Es kann individuell auch prophylaktische Mastektomie beidseits diskutiert werden, wird derzeit aber von der Krankenkasse nicht als Pflichtleistung anerkannt.
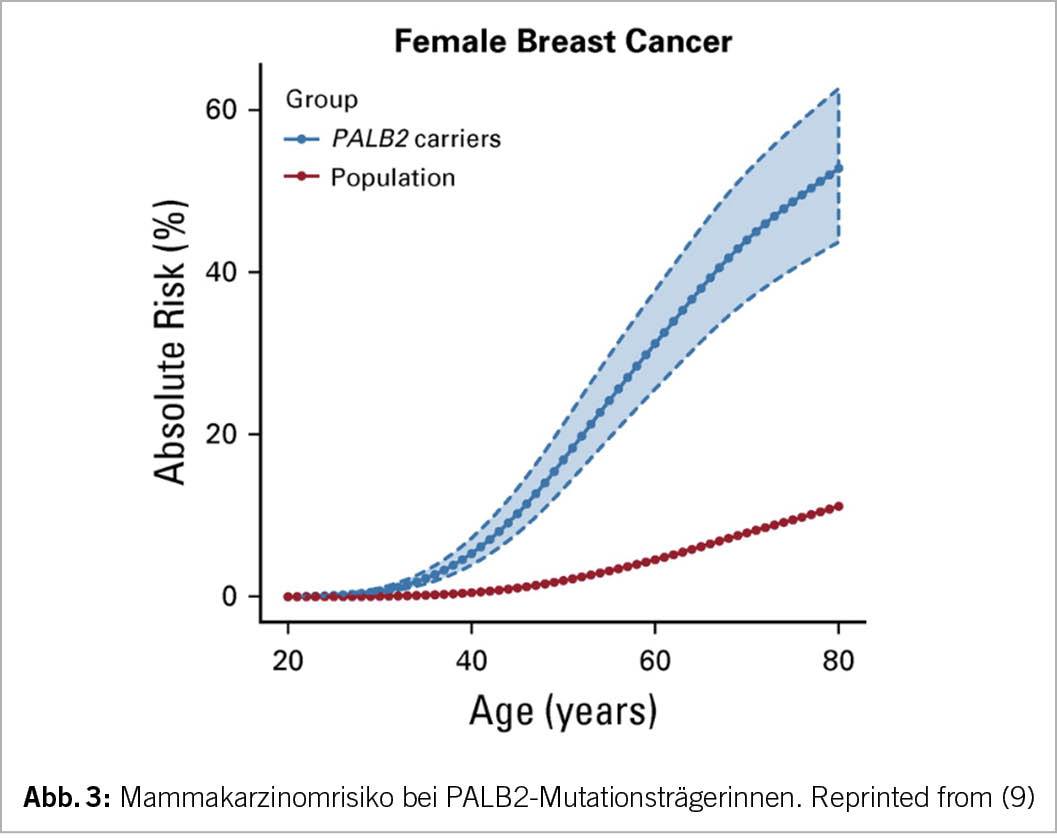
Selbstverständlich stehen PALB2-Mutationsträgerinnen auch eine intensivierte Überwachung mit Bildgebung ab jungem Alter zu. Bezüglich Ovarialkarzinom liegt derzeit leider keine effektive Früherkennungsmöglichkeit vor. Halbjährliche Kontrollen mit Vaginalultraschall und Tumormarkerverlauf CA-125 haben in Studien bisher nicht den erhofften Effekt gezeigt (10).
Deshalb wird bei hohem Lebenszeitrisikos für ein Ovarialkarzinom eine prophylaktische bilaterale Adnexektomie empfohlen (11). Für BRCA-Mutationsträgerinnen (speziell BRCA1) wird in Studien auch eine Senkung der Sterblichkeitsrate nach einer Salpingo-Oophorektomie bewiesen (12). In diesem Zusammenhang spielt auch das Patientenalter eine wichtige Rolle. Patientinnen
mit pathogener BRCA-1-Mutation sollen nach Beendung der Familienplanung im Alter von 35-40 Jahren operiert werden, während Patientinnen mit pathogener BRCA-2-Genvariante diese Operation allenfalls etwas hinauszögern können (40-45 Jahre).
Derzeit wird in der noch laufenden TUBA-Studie untersucht (13), ob ein sequentielles Vorgehen in jungem Alter zunächst einer Salpingektomie bds. und später noch einer Ovarektomie bds. eine Option unter Berücksichtigung von Nutzen und Nebenwirkungen ist. Darüber hinaus können Patientinnen mit diesen Mutationen von zielgerichteten Therapien profitieren, falls ein Tumor diagnostiziert wird. Die Inhibitoren der Polyadenosindiphosphat-Ribose-Polymerase (PARP-Inhibitoren) stellen z.B. eine sehr zukunftsträchtige therapeutische Option dar (14).
Kürzlich haben vereinzelte Studien nämlich bewiesen, dass eine Therapie mit PARP-Inhibitoren wie Olaparib, Niraparib und Talazoparib (15, 16) sowohl zu einer verminderten Progression der Grunderkrankung als auch zu einer Erhöhung des Überlebens führt.
Schlussendlich soll auch erwähnt werden, dass bei BRCA1-Mutationsträgerinnen besser auf eine hormonelle (kombinierte Ovulationshemmer) Antikonzeption verzichtet werden soll, insbesondere wenn Mammakarzinome in jungem Alter in der Familie prädominant vorkommen. In Familien mit vorwiegend Ovarialkarzinomen kann die Einnahme eines Ovulationshemmers aber protektiv sein. Die Vor- und Nachteile der verschiedenen Optionen sollen dementsprechend während der Beratung diskutiert werden (17).
Andere Genmutationen
Mutationen in anderen Genen, welche im Gen-Panel untersucht werden, sind häufig mit unterschiedlichen Tumorleiden assoziiert. Anhand der spezifisch gehäuft vorkommenden Tumorerkrankungen je nach betroffenem Gen werden entsprechende Früherkennungsprogramme empfohlen (z.B. Koloskopie, Gastroskopie und Schilddrüsensonografie). Prophylaktische operative Eingriffe werden in der Regel nicht empfohlen ausser bei Mutationen im Zusammenhang mit dem Lynchsyndrom bei Mutationen in MLH1, MSH2, MSH6 und PMS2 (prophylaktische Hysterektomie und Adnektomie nach Abschluss der Familienplanung je nach Art der Mutation und Stammbaum).
Bei pathogenen Mutationen in weniger bekannten Genen im Zusammenhang mit Tumoren stehen online Risikoeinschätzungen und eine Zusammenstellung der aktuellen Empfehlungen (NCCN, ESMO, Graffeo) zum Management online zur Verfügung (www.ask2me.ch)
Schlussfolgerung
Keimbahnmutationen in Zusammenhang mit einer erhöhten Anfälligkeit für Tumoren haben mittlerweilen auf verschiedenste Aspekte des Managements von Erkrankten und ihren Familienangehörigen einen zunehmend grossen Einfluss, wie z.B. Prävention, Screening und Behandlung. Die Beratung und Testung von Personen mit erblicher Krebsveranlagung sind in den letzten Jahren zunehmend komplexer geworden, und hochspezialisiertes Personal mit interdisziplinären Kompetenzen wird zur Beratung zunehmend benötigt.
Genetische Tests auf Keimbahnmutationen sollen bei jenen Personen in Betracht gezogen werden, bei denen sich therapeutische oder prophylaktische Konsequenzen daraus ergeben. Somit können das effektivste Management und die effizienteste Nutzung von Gesundheitsressourcen den betroffenen Familien angeboten werden.
Copyright bei Aerzteverlag medinfo AG
Universitätsspital Basel, Frauenklinik
Spitalstrasse 21, 4031 Basel
Universitätsspital Basel, Frauenklinik
Spitalstrasse 21, 4031 Basel
nicole.buerki@usb.ch
Die Autorinnen haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Arndt V, Feller A, Hauri D, Heusser R, Junker C, Kuehni C, et al. Swiss Cancer Report 2015 – Current situation and developments. Neuchâtel: Federal Statistical Office (FSO); 2016
2. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108(44):18032–7.
3. Kraemer D, Azzarello-Burri S, Steindl K, Boonsawat P, Zweier M, Dedes KJ, Joset P, Fink D, Rauch A. Prevalence of genetic susceptibility for breast and ovarian cancer in a non-cancer related study population: secondary germline findings from a Swiss single centre cohort. Swiss Med Wkly. 2019 Aug 18;149:w20092.
4. Stoll S, Unger S, Azzarello-Burri S, Chappuis P, Graffeo R, Pichert G, Röthlisberger B, Taban F, Riniker S. Update Swiss guideline for counselling and testing for predisposition to breast, ovarian, pancreatic and prostate cancer. Swiss Med Wkly. 2021 Sep 13;151:w30038.
5. Li S, Silvestri V, Leslie G, Rebbeck TR, Neuhausen SL, Hopper JL, Nielsen HR, Lee A, Yang X, McGuffog L, Parsons MT, Andrulis IL, Arnold N, Belotti M, Borg Å, Buecher B, Buys SS, Caputo SM, Chung WK, Colas C, Colonna SV, Cook J, Daly MB, de la Hoya M, de Pauw A, Delhomelle H, Eason J, Engel C, Evans DG, Faust U, Fehm TN, Fostira F, Fountzilas G, Frone M, Garcia-Barberan V, Garre P, Gauthier-Villars M, Gehrig A, Glendon G, Goldgar DE, Golmard L, Greene MH, Hahnen E, Hamann U, Hanson H, Hassan T, Hentschel J, Horvath J, Izatt L, Janavicius R, Jiao Y, John EM, Karlan BY, Kim SW, Konstantopoulou I, Kwong A, Laugé A, Lee JW, Lesueur F, Mebirouk N, Meindl A, Mouret-Fourme E, Musgrave H, Ngeow Yuen Yie J, Niederacher D, Park SK, Pedersen IS, Ramser J, Ramus SJ, Rantala J, Rashid MU, Reichl F, Ritter J, Rump A, Santamariña M, Saule C, Schmidt G, Schmutzler RK, Senter L, Shariff S, Singer CF, Southey MC, Stoppa-Lyonnet D, Sutter C, Tan Y, Teo SH, Terry MB, Thomassen M, Tischkowitz M, Toland AE, Torres D, Vega A, Wagner SA, Wang-Gohrke S, Wappenschmidt B, Weber BHF, Yannoukakos D, Spurdle AB, Easton DF, Chenevix-Trench G, Ottini L, Antoniou AC. Cancer Risks Associated With BRCA1 and BRCA2 Pathogenic Variants. J Clin Oncol. 2022;40:1529-1541.
6. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, Jervis S, van Leeuwen FE, Milne RL, Andrieu N, Goldgar DE, Terry MB, Rookus MA, Easton DF, Antoniou AC; BRCA1 and BRCA2 Cohort Consortium, McGuffog L, Evans DG, Barrowdale D, Frost D, Adlard J, Ong KR, Izatt L, Tischkowitz M, Eeles R, Davidson R, Hodgson S, Ellis S, Nogues C, Lasset C, Stoppa-Lyonnet D, Fricker JP, Faivre L, Berthet P, Hooning MJ, van der Kolk LE, Kets CM, Adank MA, John EM, Chung WK, Andrulis IL, Southey M, Daly MB, Buys SS, Osorio A, Engel C, Kast K, Schmutzler RK, Caldes T, Jakubowska A, Simard J, Friedlander ML, McLachlan SA, Machackova E, Foretova L, Tan YY, Singer CF, Olah E, Gerdes AM, Arver B, Olsson H. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA. 2017;317:2402-2416.
7. Heemskerk-Gerritsen BAM, Jager A, Koppert LB, Obdeijn AI, Collée M, Meijers-Heijboer HEJ, Jenner DJ, Oldenburg HSA, van Engelen K, de Vries J, van Asperen CJ, Devilee P, Blok MJ, Kets CM, Ausems MGEM, Seynaeve C, Rookus MA, Hooning MJ. Survival after bilateral risk-reducing mastectomy in healthy BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat. 2019;177:723-733.
8. Tung NM , Boughey JC , Pierce LJ , Robson ME , Bedrosian I , Dietz JR , et al. Management of Hereditary Breast Cancer: American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology Guideline. J Clin Oncol. 2020 Jun;38(18):2080–106
9. Yang X, Leslie G, Doroszuk A, Schneider S, Allen J, Decker B, Dunning AM, Redman J, Scarth J, Plaskocinska I, Luccarini C, Shah M, Pooley K, Dorling L, Lee A, Adank MA, Adlard J, Aittomäki K, Andrulis IL, Ang P, Barwell J, Bernstein JL, Bobolis K, Borg Å, Blomqvist C, Claes KBM, Concannon P, Cuggia A, Culver JO, Damiola F, de Pauw A, Diez O, Dolinsky JS, Domchek SM, Engel C, Evans DG, Fostira F, Garber J, Golmard L, Goode EL, Gruber SB, Hahnen E, Hake C, Heikkinen T, Hurley JE, Janavicius R, Kleibl Z, Kleiblova P, Konstantopoulou I, Kvist A, Laduca H, Lee ASG, Lesueur F, Maher ER, Mannermaa A, Manoukian S, McFarland R, McKinnon W, Meindl A, Metcalfe K, Mohd Taib NA, Moilanen J, Nathanson KL, Neuhausen S, Ng PS, Nguyen-Dumont T, Nielsen SM, Obermair F, Offit K, Olopade OI, Ottini L, Penkert J, Pylkäs K, Radice P, Ramus SJ, Rudaitis V, Side L, Silva-Smith R, Silvestri V, Skytte AB, Slavin T, Soukupova J, Tondini C, Trainer AH, Unzeitig G, Usha L, van Overeem Hansen T, Whitworth J, Wood M, Yip CH, Yoon SY, Yussuf A, Zogopoulos G, Goldgar D, Hopper JL, Chenevix-Trench G, Pharoah P, George SHL, Balmaña J, Houdayer C, James P, El-Haffaf Z, Ehrencrona H, Janatova M, Peterlongo P, Nevanlinna H, Schmutzler R, Teo SH, Robson M, Pal T, Couch F, Weitzel JN, Elliott A, Southey M, Winqvist R, Easton DF, Foulkes WD, Antoniou AC, Tischkowitz M. Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J Clin Oncol. 2020;38:674-685.
10. Grandi G, Fiocchi F, Cortesi L, Toss A, Boselli F, Sammarini M, Sighinolfi G, Facchinetti F. The challenging screen detection of ovarian cancer in BRCA mutation carriers adhering to a 6-month follow-up program: results from a 6-years surveillance. Menopause. 2021;29:63-72.
11. Lewis KE, Lu KH, Klimczak AM, Mok SC. Recommendations and Choices for BRCA Mutation Carriers at Risk for Ovarian Cancer: A Complicated Decision. Cancers (Basel). 2018;10:57.
12. Marchetti C, De Felice F, Palaia I, Perniola G, Musella A, Musio D, Muzii L, Tombolini V, Panici PB. Risk-reducing salpingo-oophorectomy: a meta-analysis on impact on ovarian cancer risk and all cause mortality in BRCA 1 and BRCA 2 mutation carriers. BMC Womens Health. 2014;14:150.
13. Steenbeek MP, Harmsen MG, Hoogerbrugge N, de Jong MA, Maas AHEM, Prins JB, Bulten J, Teerenstra S, van Bommel MHD, van Doorn HC, Mourits MJE, van Beurden M, Zweemer RP, Gaarenstroom KN, Slangen BFM, Brood-van Zanten MMA, Vos MC, Piek JMJ, van Lonkhuijzen LRCW, Apperloo MJA, Coppus SFPJ, Massuger LFAG, IntHout J, Hermens RPMG, de Hullu JA. Association of Salpingectomy With Delayed Oophorectomy Versus Salpingo-oophorectomy With Quality of Life in BRCA1/2 Pathogenic Variant Carriers: A Nonrandomized Controlled Trial. JAMA Oncol. 2021;7:1203-1212.
14. Moore K , Colombo N , Scambia G , Kim BG , Oaknin A , Friedlander M , et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N Engl J Med. 2018 Dec;379(26):2495–505.
15. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, Conte P. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017;377:523-533.
16. Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, Roché H, Im YH, Quek RGW, Markova D, Tudor IC, Hannah AL, Eiermann W, Blum JL. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N Engl J Med. 2018;379:753-763.
17. Schrijver LH, Mooij TM, Pijpe A, Sonke GS, Mourits MJE, Andrieu N, Antoniou AC, Easton DF, Engel C, Goldgar D, John EM, Kast K, Milne RL, Olsson H, Phillips KA, Terry MB, Hopper JL, van Leeuwen FE, Rookus MA. Oral Contraceptive Use in BRCA1 and BRCA2 Mutation Carriers: Absolute Cancer Risks and Benefits. J Natl Cancer Inst. 2022 Apr 11;114(4):540-552.


info@gynäkologie
- Vol. 13
- Ausgabe 1
- Februar 2023