
Symposiums, Prof. Corinna Brunckhorst und Prof. Firat Duru, beide von der Klinik für Kardiologie des Universitätsspitals Zürich, konnten über 90 hochkarätige Experten aus aller Welt im Hotel Belvoir in Rüschlikon begrüssen.

Ziel des diesjährigen Symposiums war es, die Grundlagen für das Consensus Dokument über die Genotyp basierten Guidelines zur Arrhythmogenen Kardiomyopathie (ARVC) zu schaffen. Das Management soll in Zukunft patientenspezifischer werden und dementsprechend war ein wichtiger Teil des Symposiums der genbasierten Diagnostik und Therapie gewidmet. Das dreitägige Symposium gliederte sich in 6 Sitzungen. Der erste Tag beinhaltete einen allgemeinen Überblick über die Pathologie, Pathophysiologie und Krankheitsmechanismen, die genetische Basis der Erkrankung, weiterhin über die Elektrokardiographie, Echokardiographie, Magnetresonanztomographie, Risikostratifizierung und den Einfluss von körperlicher Aktivität sowie die Präsentation der arrhythmogenen Kardiomyopathie im jungen Alter mit speziellen Betrachtungen für die pädiatrische Population.
Der zweite Tag beinhaltete zwei Sitzungen zu Genotypen: Plakophilin-2 (PKP2) Kardiomyopathie, Desmoglein-2 (DSG2/Desmocollin-2 (DSC2) Kardiomyopathie, Desmoplakin (DSP) Kardiomyopathie sowie Phospholamban (PLN) und Transmembranprotein 43 (TMEM43) Kardiomyopathie und am Nachmittag Plakoglobin (JUP), Desmin (DES), Filamin-C (FLNC) und Cadherin-2 (CHH2) Kardiomyopathie Anschliessend wurden die verschiedenen Therapiestrategien, wie Antiarrhythmika, Herzinsuffizenztherapien, Device Indikationen und die Katheterablation diskutiert.
Der letzte Tag begann mit Vorträgen zu immunsuppressiver Therapie und Gentherapie und endete mit der Roadmap zum Konsensus-Dokument und der Planung der nächsten Schritte.
Der folgende Bericht enthält einen Auszug aus den zahlreichen Präsentationen.
Allgemeine Sessionen I
Pathologie der Arrhythmogenen Kardiomyopathie
Prof. Christina Basso, Padua, und Prof. Jeffrey Saffitz, Boston, erinnerten zunächst an eine erste Publikation zum Thema aus dem Jahre 1982 (Marcus FI et al. Right ventricular dysplasia, a report of 24 adult cases. Circulation 1982;65:384-98), in der 24 Fälle von Erwachsenen mit ventrikulärer Dysplasie untersucht wurden. Die Autoren kamen zu folgendem Schluss: Die rechtsventrikuläre Dysplasie ist durch eine Anomalie in der Entwicklung eines Teils der rechtsventrikulären Muskulatur gekennzeichnet. Bei Patienten mit rechtsventrikulärer Dysplasie können ventrikuläre Tachykardien, supraventrikuläre Arrhythmien, Rechtsherzinsuffizienz oder asymptomatische Kardiomegalie auftreten.

Bei dem Versuch, die arrhythmogene rechtsventrikuläre Kardiomyopathie klinisch zu diagnostizieren, wurde eine Vielzahl von zum Teil aufwendigen bildgebenden Verfahren eingesetzt. Keines dieser Verfahren hat bisher so überzeugende Ergebnisse geliefert, dass es uneingeschränkt als Referenzverfahren angesehen werden kann. Die Schwierigkeiten in der klinischen Diagnostik der Erkrankung spiegeln sich auch in dem Vorschlag wider, die arrhythmogene rechtsventrikuläre Dysplasie/Kardiomyopathie anhand von Haupt- und Nebenkriterien zu diagnostizieren, ohne die Art der bildgebenden Verfahren zu berücksichtigen. Strukturelle, histologische, elektrokardiographische Arrhythmien und genetische Faktoren dienen als Arbeitsrahmen für eine bessere Erkennung dieser Erkrankung. Dies sollte zu einer besseren Darstellung des Stammbaums, zur Identifizierung der verantwortlichen Gene und zu einem besseren Verständnis des natürlichen Verlaufs führen. Diese Ansätze ermöglichen entweder eine präzisere klinisch-pathologische Diagnose oder im Idealfall eine Diagnose auf der Grundlage einer genetischen Anomalie.
Genetische Basis der ACM und genetische Testung
Die Themen von Prof. Peter van Tintelen, Amsterdam und Prof. Cindy James, Baltimore waren:

Genetische Basis der ACM / Rationale
• ACM-Gene
• Prinzipien der ACM-Vererbung
Genetische Testung
• Strategie für Genanalysen und Testauswahl
• Auswertung der Ergebnisse genetischer Tests
• genetische Beratung
• besondere Überlegungen – Kinder, reproduktive Entscheidungsfindung
ACM-Definition und phänotypische Klassifikation
Abb. 1
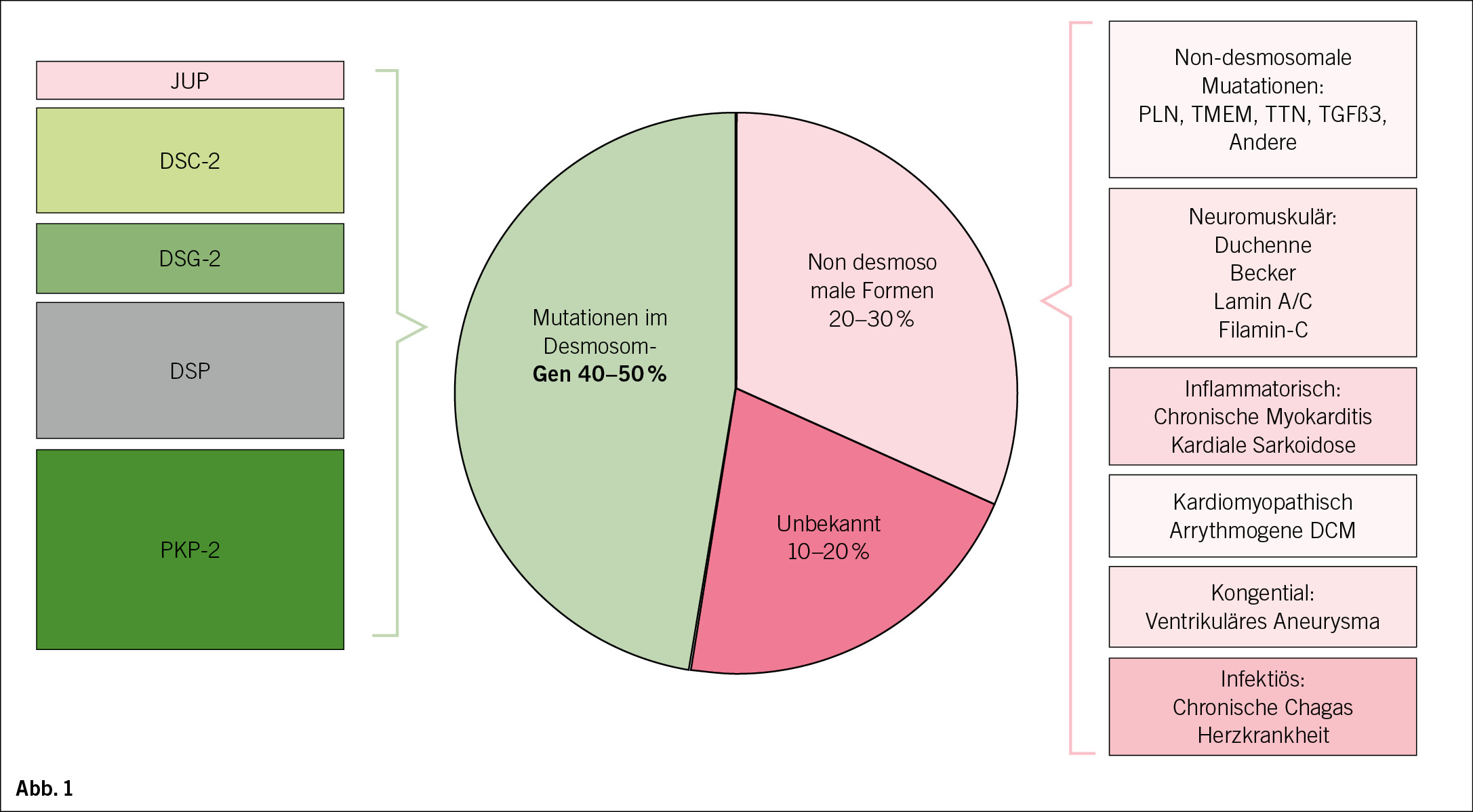
ACM/ARVC-assoziierte Gene
Die Referenten präsentieren eine internationale, evidenzbasierte Neubewertung von Genen, die mit ARVC in Verbindung gebracht werden, unter Verwendung des ClinGen Framework (Cynthia James et al Circ Genome Precis Med 2021 Apr 8.doi 10.1161/CIRCGEN .12000 3273).Bei der Verwendung des Ansatzes der Clinical Genome Resource zur Gen-Krankheits-Kuration wurden nur bei 8 Genen (PKP2, DSP, DSG2, DSC2, JUP, TMEM43, PLN und DES) eindeutige oder mäßige Hinweise auf ARVC gefunden, und diese Gene machten fast alle pathogenen/wahrscheinlich pathogenen ARVC-Varianten in ClinVar aus. Daher sollten nur pathogene/wahrscheinlich pathogene Varianten in diesen 8 Genen ein Hauptkriterium für die ARVC-Diagnose darstellen. Pathogene/wahrscheinlich pathogene Varianten, die in anderen Genen bei einem Patienten identifiziert werden, sollten eine weitere Phänotypisierung veranlassen, da Varianten in vielen dieser Gene mit anderen kardiovaskulären Erkrankungen in Verbindung stehen.
Definitive Gene sind DSP, PKP2, DSC2, DSG2, JUP, TMEM43. Moderate Gene: DES, PLN, umfasst auch CDH2 und FLNC.
ACM-Vererbung – die Grundlagen
In der Regel autosomal-dominant mit reduzierter altersbedingter Penetranz und variabler Expressivität. De-novo-Varianten sind selten. Es gibt Gen- und Genotyp-spezifische Ausnahmen.
James et al (Eur Heart J 2020;41:1393-1400) beschrieben ein neues Schwellenwertmodell für die ACM-Vererbung, bei dem mehrere Faktoren, einschließlich pathogener Varianten in bekannten ACM-Genen, genetischer Modifikatoren und Umweltexpositionen, insbesondere körperliche Aktivität, erforderlich sind, um einen Schwellenwert für die Krankheitsexpression zu erreichen. Die Autoren überprüften auch Best Practices für die Integration von Genetik -einschließlich jüngster Entdeckungen – in die Pflege von ACM-Familien und betonen den Nutzen des Genotyps sowohl für das Management betroffener Personen als auch für prädiktive Tests bei Familienmitgliedern.
Komplexität – multiple PLP- Varianten
Das Vorhandensein mehrerer pathogener Varianten in desmosomalen Genen (DSC2, DSG2, DSP, JUP und PKPs) bei Patienten mit ARVC wurde mit einem schweren Phänotyp in Verbindung gebracht. Die Pathogenität der Varianten wird jedoch häufig neu klassifiziert, was zu einer veränderten klinischen Risikovorhersage führen kann. In einer letztjährigen Publikation (Nagyova E et al. J Cardiovas Trans Res 2023) wurde die Erfassung, Reklassifizierung und Korrelation der klinischen Ergebnisse für die bisher grösste Serie von ARVC-Patienten, die mehrere desmosomale pathogene Varianten tragen (n = 331) zusammengestellt. Nach der Reklassifizierung bleiben nur 29% der Patienten Träger von zwei (Wahrscheinlich) pathogenen Varianten. Sie erreichten den zusammengesetzten Endpunkt (ventrikulärer Arrhythmien, Herzversagen durch Tod) deutlich früher als Patienten mit einer oder keiner verbleibenden reklassifizierten Variante. Die regelmässige Reklassifizierung von Varianten trägt zu einer genaueren Risikostratifizierung und einer anschliessenden klinischen Behandlungsstrategie bei.
Gentest:
Zunächst eine Familien- und Gentestanamnese durchführen.
Gentest-Geschichte:
• Hat der Patient einen vorherigen Gentest gehabt?
• Wurde eine ähnliche genetische Ursache bereits identifiziert?
• Wenn ein früherer Test negativ war, was war die verwendete Methodologie-Generationsstammbaum und die genetische Abdeckung?
Familienanamnese – Erhalte einen Dreigenerationen-Stammbaum
• Ist eine P/LP Variante bereits in der Familie bekannt?
• Gibt es eine bekannte genetische Diagnose in der Familie?
• Gibt es ein lebendes Familienmitglied mit signifikant jüngerer oder schwerer Krankheit?
• Verlasse Dich nicht auf Resultate von «Freizeit-Gentests»
ACM-Gentest – Methodik und Testspektrum
Gentest für präsymptomatische Testung
• LP/P Variante beim Probanden →präsymptomatisch (Kaskade) genetische Testung, Beratung
• (nur für die entsprechende Variante)
Gentest für Segregationsanalyse-VUS beim Probanden → VUS-Testung bei den Eltern/betroffenen Verwandten.
ACM Genetische Testung
• Analytische Validität (niemals 100%) Regeln der nationalen Verbände und Vorschriften
Kaskadentest
Es wird ACM -Probanden empfohlen allen jugendlichen und erwachsenen Verwandten ersten Grades eines G+ ACM betroffenen Verwandten Gentests anzubieten.
Molekulare Autopsie
Klinische Beratung:
Teste wenn möglich einen nicht betroffenen Verwandten
DNA- Bank sollte Betroffenen angeboten werden. Beratung über die relativ geringe Wahrscheinlichkeit eines positiven Ergebnis. Idealerweise Konzentration auf wahrscheinliche Kandidaten-Gene, von denen bekannt ist, dass sie in kausalem Zusammenhang mit dem vermuteten Phänotyp stehen.
Klinischer Kontext des Gen-erst-Ansatzes bei ACM
Probanden – heterogene, existierende Guidelines
Molekulare Autopsie – sich abzeichnende Guidelines
Kaskaden genetische Testung (Verwandte – Konsistente starke fachliche Empfehlungen für Kaskaden-Gentests)
Zuerst Gen/sekundäre Findungskohorten – ACM Gene werden als Sekundärbefund empfohlen, wenn P/LP-Varianten entdeckt werden.
Genetische Beratung
Klinische Beratung; Die genetische Beratung ist ein integraler Bestandteil des ACM-Gentestverfahrens. Sie sollte sowohl die Bereitstellung von Informationen als auch psychosoziale Unterstützung umfassen.
Pädiatrische Gentests
Probanden /symptomatische Verwandte: Patienten, bei denen die Symptome in der Pädiatrie festgestellt werden – vor allem Probanden – haben oft einen schwereren Krankheitsverlauf. Gentests bei klinisch betroffenen pädiatrischen und adoleszenten Probanden werden empfohlen.
Asymptomatische Verwandte
Genetische Kaskadentests in der Pädiatrie können in Absprache mit dem Patienten und seiner Familie angebracht sein, um eine Entscheidung für ein kardiales Screening und eine bestimmte Lebensweise zu treffen. Das Testalter sollte sich nach dem Genotyp und der Familienanamnese richten und die Werte und Präferenzen der Familie einbeziehen.
Alle
Genetische Untersuchungen bei Kindern und Jugendlichen sollten eine umfassende genetische Bewertung vor dem Test beinhalten, um sowohl den medizinischen als auch den psychosozialen Auswirkungen der Testergebnisse zu besprechen.
Reproduktive Entscheidungsfindung
Klinische Beratung: Eine genetische Beratung sollte denjenigen angeboten werden, die über die Vererbung von ACM oder pränatale Gentests und reproduktive Optionen für die Familienplanung sprechen möchten. Verfügbarkeit und Leitlinien für diese Optionen sind eine wichtige Voraussetzung für die Entscheidungsfindung.
riesen@medinfo-verlag.ch
