Kardiomyopathien sind Herzmuskelerkrankungen mit strukturellen und funktionellen Anomalien des Herzmuskels. In dieser Übersicht werden pathophysiologische Aspekte, die klinische Manifestation, die Diagnosestellung, die Risikostratifizierung und die neusten therapeutischen Konzepte der drei häufigsten Kardiomyopathieformen – hypertrophe Kardiomyopathie (HCM), dilatative Kardiomyopathie (DCM) und arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC) – aufgezeigt. Zusätzlich berichten wir über die in den Richtlinien für das Management von Kardiomyopathien der Europäischen Gesellschaft für Kardiologie (ESC) von 2023 neu definierte Entität der nicht-dilatierten linksventrikulären Kardiomyopathie (NDLVC).
Cardiomyopathies are usually hereditary conditions of the heart muscle including structural and functional abnormalities. In this overview we cover pathophysiological aspects, clinical manifestations, diagnostics, risk stratification and the newest therapeutic concepts of the three most common forms of cardiomyopathy: hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and arrhythmogenic cardiomyopathy (ARVC). Furthermore, we report on the newly described for of non-dilated left ventricular cardiomyopathy (NDLVC).
Key Words: hypertrophic cardiomyopathy, dilated cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, non-dilated left ventricular cardiomyopathy.
Einleitung
Kardiomyopathien sind Herzmuskelerkrankungen, bei denen der Herzmuskel strukturell und funktionell auffällig ist, ohne dass eine erkennbare Grunderkrankung vorliegt (1).
Obwohl viele dieser Erkrankungen genetisch bedingt sind, ist eine rein genetische Klassifikation in der klinischen Praxis weder sinnvoll noch möglich (2). Es ist wichtig zu erkennen, dass verschiedene Phänotypen von Kardiomyopathien innerhalb einer Familie koexistieren können und dass der Progress der Erkrankung beim einzelnen Patienten von einem Kardiomyopathie-Phänotyp zu einem anderen führen kann (3). Daher werden die Kardiomyopathien weiterhin nach morphologischen und funktionellen Kriterien eingeteilt, wobei es zu Überschneidungen zwischen den verschiedenen Formen kommen kann (2). Es ist daher nicht möglich, ein einziges Klassifikationsschema für alle möglichen Ursachen und Krankheitsbilder zu erstellen. So wurde in den ESC Guidelines 2023 die bestehende klinische Klassifikation aktualisiert, um neue phänotypische Beschreibungen aufzunehmen und die Terminologie zu vereinfachen, während sie gleichzeitig einen konzeptionellen Rahmen für Diagnose und Behandlung bietet (3). Es wird jedoch empfohlen, bei der Nomenklatur und Diagnose der Erkrankung nach dem bei der Präsentation vorherrschenden kardialen Phänotyp vorzugehen (3).
In diesem Artikel werden die drei häufigsten Formen der Kardiomyopathien beschrieben: die hypertrophe Kardiomyopathie (HCM), die dilatative Kardiomyopathie (DCM) und die arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC) sowie die neu definierte Entität der nicht-dilatierten linksventrikulären Kardiomyopathie (NDLVC).
Hypertrophe Kardiomyopathie (HCM)
Definition
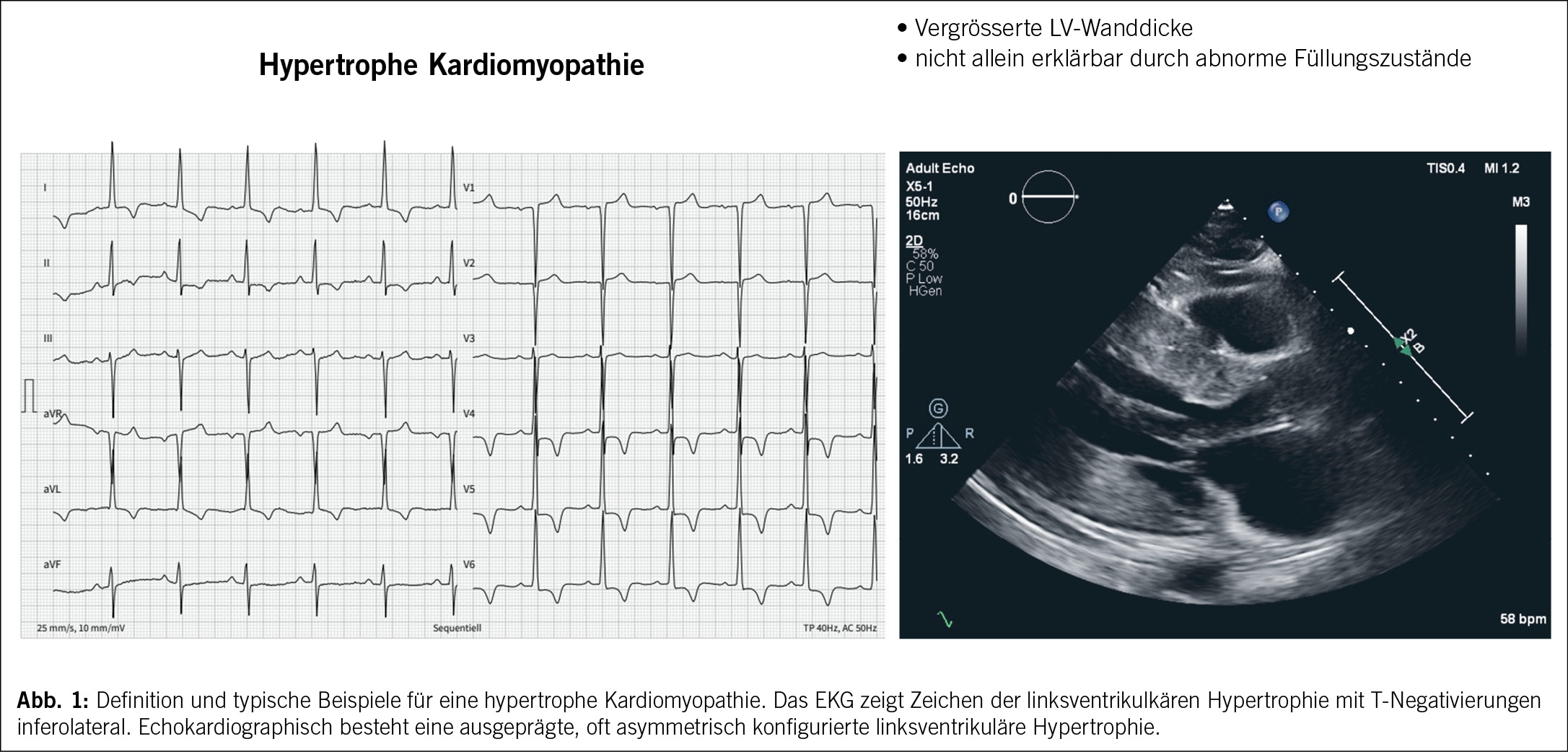
Die HCM ist definiert als das Vorliegen einer erhöhten linksventrikulären Wanddicke (mit oder ohne rechtsventrikulärer Hypertrophie) oder Masse, die nicht allein durch abnormale linksventrikuläre Füllungszustände erklärt werden kann (3). (Abb. 1)
Die HCM ist eine relativ häufige genetisch bedingte Herzerkrankung, die in der Regel autosomal dominant vererbt wird und weltweit in allen Bevölkerungsgruppen vorkommt (1).
Pathogenese
Die Annahme einer morphologischen Krankheitsdefinition impliziert eine Reihe unterschiedlicher Ätiologien, die zu einer hypertrophen Kardiomyopathie führen können. Ca. 60 % aller Fälle sind auf Varianten in Genen für kardiale Sarkomerproteine, insbesondere in der schweren β-Myosinkette (MYH7) und dem Myosin-bindenden Protein C (MYPBC3) zurückzuführen (1). Erbliche Stoffwechselstörungen wie Morbus Fabry, infiltrative Speichererkrankungen wie Amyloidose, neuromuskuläre und endokrine Pathologien, Mitochondriopathien und genetische Syndrome verursachen etwa 5-10 % der Fälle. Für einige von ihnen gibt es kurative Optionen (4).
Die klassische sarkomerische Form der HCM ist typischerweise eine monogenetische Erkrankung. Das typische Erscheinungsbild mit diastolischer Dysfunktion und ventrikulären Arrhythmien wird durch die Dysfunktion des sarkomerischen Proteins und das dadurch verursachte adverse Remodelling verursacht. Auf zellulärer Ebene sind vor allem Veränderungen der Aktin-Myosin-Interaktionen und der Ca2+-Sensitivität zu verzeichnen. Für das Verständnis der aktuellen Therapieoptionen ist hier vor allem relevant, dass HCM-verursachende Genvarianten zu vermehrten kardialen Aktin-Myosin-Querbrücken, welche wiederum für hyperdynamische Kontraktionen und gesteigerten Energieverbrauch verantwortlich sind (1, 5, 6).
Klinik
Das klinische Bild der HCM kann sehr unterschiedlich sein. Es kann als Zufallsbefund bei einer Vorsorgeuntersuchung entdeckt werden oder sich als Auftreten neuer Symptome wie Dyspnoe, Angina pectoris, Palpitationen und Synkopen oder sich im schlimmsten Fall primär durch einen Herzstillstand zeigen (5).
Diagnose
Bei Erwachsenen kann die klinische Diagnose einer HCM durch eine maximale enddiastolische Wanddicke von ≥ 15 mm in einem oder mehreren linksventrikulären Segmenten bei Fehlen anderer Ursachen der Hypertrophie, gemessen mittels Echokardiographie, Magnetresonanztomographie des Herzens oder Computertomographie definiert werden (1).
Wie bei allen Kardiomyopathien berichten die Patienten sehr unterschiedliche Beschwerden wie Dyspnoe, Thoraxschmerzen, Palpitationen, Synkopen oder Leistungsintoleranz, viele Patienten werden jedoch auch als Zufallsbefund diagnostiziert (3). Ein wichtiger Teil der Anamnese ist die Analyse des Stammbaums über drei Generationen bezüglich Herzerkrankungen, plötzlichem Herztod oder unklaren Unfällen, welche auf Synkopen hinführen könnten. Das 12-Kanal-EKG ist häufig pathologisch und kann daher ein hilfreiches Instrument sein, da Zeichen einer linksventrikulären Hypertrophie, ST-, T- sowie Q-Wellenanomalien nachgewiesen werden können. Ebenso wichtig ist das 24-Stunden-EKG, mit dem das Vorliegen nicht anhaltenden ventrikulärer Tachykardien dokumentiert wird und das Risiko eines plötzlichen Herztodes berechnet werden kann. Zusätzlich kann Vorhofflimmern erkannt und damit das Schlaganfallrisiko bestimmt werden (7).
Die Echokardiographie ist das wichtigste diagnostische Mittel bei der Abklärung einer HCM. Sie kann die Diagnose stellen, die Ursache der Symptome identifizieren und ist somit nützlich für das Therapiemanagement und schliesslich für die Risikostratifizierung des plötzlichen Herztodes (7).
Risikostratifizierung
Da die HCM als eine relevante Ursache für den plötzlichen Herztod vor allem bei jungen Menschen gilt, ist es ein Ziel, das Risiko für das Auftreten maligner Arrhythmien abzuschätzen und ein geeignetes Risikomodell zur Abschätzung der Häufigkeit zukünftiger kardialer Ereignisse zu finden, um diese durch die Implantation eines ICDs zu verhindern.
Die Empfehlung, die Überlebenden von SCD mit einem ICD zu versorgen, wurde bereits vor Jahren ausgesprochen; die Debatte über die Primärprophylaxe einer ICD-Implantation in der HCM-Population ist jedoch eine schwierigere Frage. Im Jahr 2014 haben O’Mahony et al. ein Risikoprädiktionsmodell, das nun von der ESC zur Stratifizierung des individuellen Risikos für SCD bei HCM und zur Unterstützung der Entscheidung für eine ICD-Implantation dient, vorgestellt (8). Da das Risiko für tödliche Arrhythmien über einen langen Zeitraum bestehen bleiben kann, ist es wichtig, das Risiko des Patienten regelmässig neu zu bewerten (1).
Management
Das Ziel der HCM-Behandlung besteht darin, die Symptome der Patienten (Angina Pectoris, Herzinsuffizienz, Synkopen, Palpitationen usw.) zu lindern und ein Fortschreiten der Krankheit sowie schwerwiegende kardiovaskuläre Komplikationen und den plötzlichen Herztod zu verhindern (1).
Das Hauptmerkmal, das bei HCM-Patienten zu Symptomen führt, ist das Vorhandensein einer linksventrikulären Ausflusstraktobstruktion, allgemein definiert als ein Doppler-Spitzengradient im linksventrikulären Ausflusstrakt (LVOT) von ≥ 30 mmHg (5).
Die Erstlinientherapie in dieser Situation umfasst nicht-vasodilatierende β-Blocker und bei Unverträglichkeit oder Unwirksamkeit Nicht-Dihydropyridin-Kalziumkanalblocker. Zusätzlich kann bei schlechtem klinischem Ansprechen das Antiarrhythmikum Disopyramid verabreicht werden (1).
Kürzlich wurde eine neue vielversprechende Therapie mit dem ersten kardialen Myosin-Inhibitor zugelassen. Mavacamten ist ein reversibler kardialer Myosin-Adenosintriphosphatase (ATPase)-Hemmer, der die Bildung von Aktin-Myosin-Kreuzbrücken reduziert und dadurch die Kontraktilität verringert und den Energiehaushalt des Herzmuskels verbessert. In der kürzlich veröffentlichten klinischen Studie zur Bewertung von Mavacamten bei Erwachsenen mit symptomatischer obstruktiver hypertropher Kardiomyopathie (EXPLORER-HCM), reduzierte Mavacamten den Gradienten des linksventrikulären Ausflusstrakts (LVOT) und verbesserte die körperliche Leistungsfähigkeit im Vergleich zu Placebo bei Patienten mit HCM und symptomatischer LVOT (NYHA II-III und EF >55%); 27% der Patienten, die Mavacamten erhielten, hatten eine Reduzierung des LVOT-Gradienten auf <30 mmHg und verbesserten sich in die NYHA-Klasse I (9). Diese Therapie hat bereits Eingang in die aktuellen Richtlinien gefunden (10).
Für Patienten mit persistierenden Symptomen trotz medikamentöser Therapie stehen invasive Optionen wie die Septum-Myektomie oder die Septum-Alkohol-Ablation in spezialisierten Zentren zur Verfügung. Die Therapie von HCM-Patienten mit Herzinsuffizienz (LVEF < 50 %) richtet sich nach den etablierten Leitlinien zur Herzinsuffizienz (1).
Vorhofflimmern ist die häufigste persistierende Arrhythmie bei HCM-Patienten, die bei etwa 20% der Patienten vorkommt und mit einer hohen Inzidenz thromboembolischer Ereignisse assoziiert ist. Eine orale Antikoagulation sollte daher bei jedem Patienten mit HCM und paroxysmalem, persistierendem oder permanentem Vorhofflimmern empfohlen werden, unabhängig vom CHA2DS2Vasc-Score, es sei denn, es besteht eine strenge Kontraindikation (3, 11).
Erwachsene Patienten mit HCM können auch eine atherosklerotische Koronararterienerkrankung (KHK) entwickeln. Die Berichte über die Prävalenz der KHK bei HCM variieren, aber bei bis zu 20 % der HCM-Patienten wurde eine koexistierende KHK festgestellt. Der Befund einer schweren KHK bei HCM-Patienten ist im Vergleich zu Patienten ohne KHK oder mit leichter bis mittelschwerer KHK mit einem reduzierten Gesamtüberleben, einem reduzierten Überleben ohne SCD assoziiert. Dieser Befund kann daher als zusätzlicher prognostischer Faktor bei der Beurteilung von Patienten mit HCM herangezogen werden (12).
Dilatative Kardiomyopathie
Definition
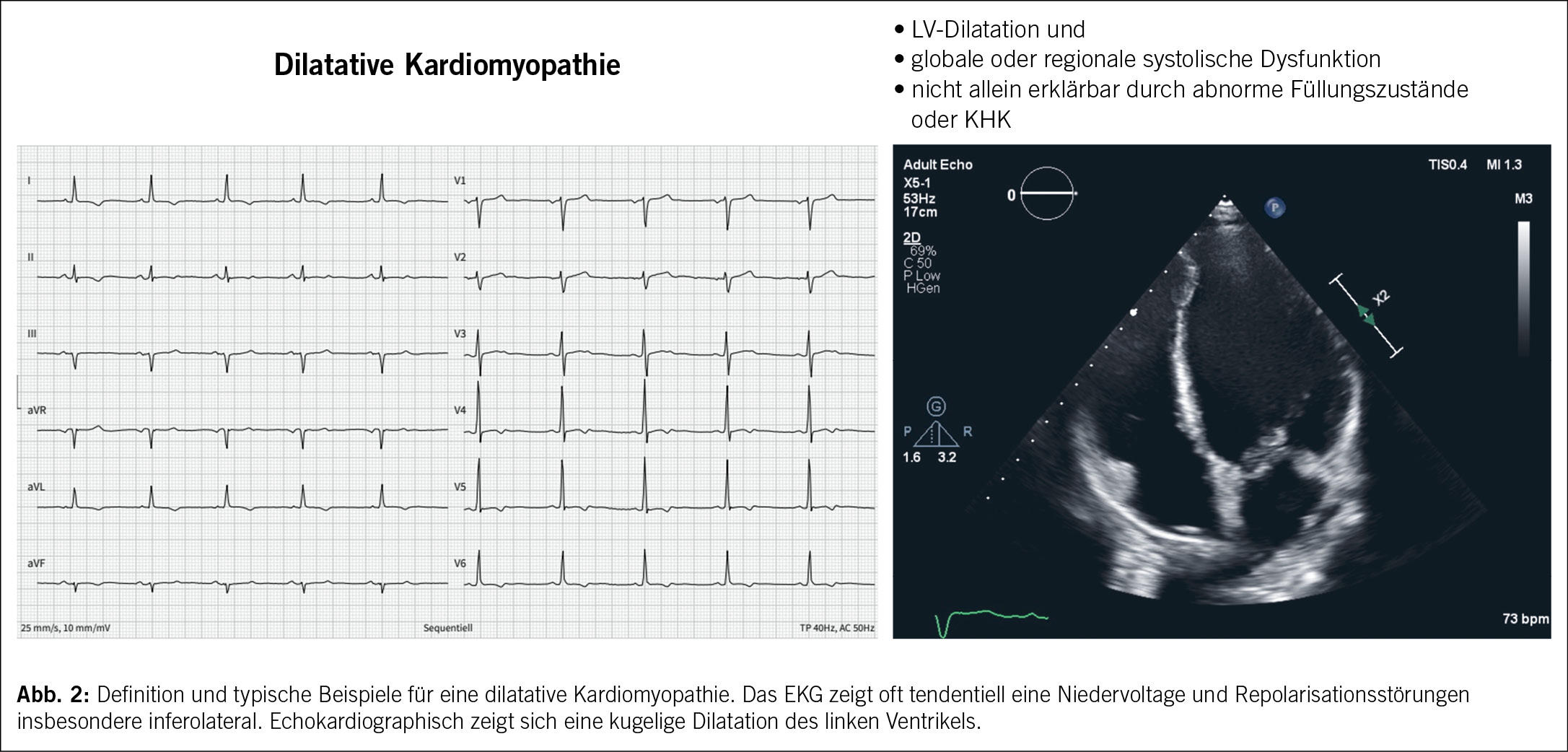
Die dilatative Kardiomyopathie (DCM) ist definiert als linksventrikuläre oder biventrikuläre Dilatation sowie systolische Dysfunktion bei Fehlen einer ischämischen, hypertensiven oder kongenitalen Herzerkrankung (1). Die ESC klassifiziert die Kardiomyopathien als familiär oder nicht-familiär, je nachdem, ob ein genetisches Substrat vorliegt oder nicht (3). (Abb. 2)
Pathogenese
Es ist bekannt, dass DCM durch eine Reihe von Prozessen verursacht werden kann. Diese wird in der klinischen Praxis gerne als «nicht-ischämische Kardiomyopathie» bezeichnet. Diese Bezeichnung entspricht jedoch nicht dem heutigen Kenntnisstand, da DCM eine Familie von Krankheiten darstellt, die durch komplexe Wechselwirkungen zwischen Umwelt und genetischer Prädisposition gekennzeichnet sind (13). Selbst in Fällen mit scheinbar eindeutiger Ursache muss zusätzlich von einer genetischen Ursache ausgegangen werden. Patienten mit alkoholbedingter DCM weisen z.B. im Vergleich zu gesunden Probanden und Bevölkerungskontrollen signifikant häufiger seltene, proteinalterierende genetische Varianten auf (14). Bei bis zu 50% der Patienten mit DCM kann eine pathogene Genvariante gefunden werden (15). Auch wenn das Wissen um die genetische Grundlage der Erkrankung in der klinischen Praxis «akademisch» erscheinen mag, kann es für den Patienten von grosser Bedeutung sein, nicht nur, um ein Kaskadenscreening bei Familienmitgliedern anzubieten, sondern auch für die Risikoabschätzung von Patienten, da das Risiko für SCD bei DCM-Patienten mit einer desmosomalen oder Lamin A/C-Genvariante deutlich erhöht ist (15). Eine Besonderheit der DCM ist das breite Spektrum möglicher Ätiologien, die einen gemeinsamen pathogenetischen Weg zur Herzschädigung implizieren, unabhängig davon, ob es sich um eine umweltbedingte oder genetische Ursache handelt, die eine Entzündung auslöst und eine Infiltration von Immunzellen verursacht, um das geschädigte Myokard zu reparieren (1).
Klinik
Die klinischen Merkmale eines Patienten mit DCM hängen hauptsächlich mit Symptomen der Herzinsuffizienz zusammen, d. h. Dyspnoe, Müdigkeit und Leistungsintoleranz, Beinödeme, pulmonale Stauung oder durch Herzrhythmusstörungen, wie Herzklopfen, Schwindel oder Synkopen. Das Ausmass dieser Symptome hängt von der Schwere der links- oder biventrikulären Dysfunktion ab, und das Auftreten kann akut, subakut oder chronisch sein und unterschiedliche Ursachen haben (16). Zusätzlich können Befunde auftreten, die Hinweise auf die genetische Grunderkrankung liefern können, wie z.B. Muskelbeschwerden (3).
Diagnostik
Die diagnostischen Kriterien basieren auf der kardialen Bildgebung, in erster Linie auf der Echokardiographie zum Nachweis eines enddiastolischen linksventrikulären Volumens oder Durchmesser > 2 SD vom Normbereich, korrigiert für Alter und Körperoberfläche sowie einer Ejektionsfraktion < 50%. Eine Koronarangiographie kann zum Ausschluss einer begleitenden koronaren Herzkrankheit durchgeführt werden. Ein MRT des Herzens kann die Dilatation bestätigen und ist entscheidend für die Beurteilung von Ödemen/Fibrosen durch spätes Gadolinium-Enhancement, das stark auf eine Entzündung hinweist und somit für die prognostische Stratifizierung nützlich ist (17). Die Myokardbiopsie kann bei Verdacht auf eine Speicherkrankheit, bei infiltrativen Prozessen und bei möglicher entzündlicher Kardiomyopathie eingesetzt werden (1).
Das breite Spektrum unterschiedlicher Ursachen erfordert aufwendige Untersuchungen, um das phänotypische Bild der DCM von der tatsächlichen Pathologie zu unterscheiden, da die Identifizierung einer spezifischen Ätiologie eine krankheitsspezifische Behandlung ermöglichen oder auf die Notwendigkeit eines Familienscreenings hinweisen und Informationen über die Prognose liefern kann (1).
Eine detaillierte Familienanamnese (bis zur dritten Generation) und eine Anamnese der Noxenexposition, Blutuntersuchung auf Eisenspeicher, Nierenfunktion, Elektrolyte, Kreatinkinase, CRP, Schilddrüsenfunktion und 12-Kanal-EKG ist unerlässlich (1).
Risikostratifizierung
Die Vorhersage von SCD ist ein schwieriger Aspekt der klinischen Versorgung von Patienten mit DCM. Implantierbare Kardioverter-Defibrillatoren (ICD) sind wirksam bei der Behandlung lebensbedrohlicher ventrikulärer Arrhythmien und bei der Prävention des plötzlichen Herztods, sind aber auch mit Komplikationen verbunden, insbesondere bei jungen Patienten, die im Laufe ihres Lebens die ICDs mehrmals ausgetauscht werden müssen (3). In der Sekundärprävention ist entsprechend die Empfehlung zur ICD-Implantation unumstritten (3).
In der Primärprävention besteht bei symptomatischen Patienten mit einer LVEF von ≤ 35% eine Klasse IIa-Indikation zur ICD-Implantation (3). Bei Patienten mit einer LVEF von > 35% müssen aufgrund der vielfältigen Ätiologie der Erkrankung zusätzliche Risikofaktoren in Betracht gezogen werden (3). Daher ist es notwendig, nach Anzeichen für Formen der Krankheit zu suchen, die ein höheres arrhythmogenes Potenzial aufweisen. Red Flags sind genetische Erkrankungen, insbesondere hochgradig arrhythmogene Formen wie Laminopathien, schwere ventrikuläre Arrhythmien, plötzlicher Herztod in der Familienanamnese, Skelettmuskelbeteiligung, EKG-Anomalien und eine posterolaterale Akinesie in der Echokardiographie. In diesen Fällen oder bei eindeutiger Familienanamnese wird eine genetische Untersuchung empfohlen, um Varianten mit erhöhtem Risiko zu erkennen (18).
Verschiedene Studien weisen darauf hin, dass der Genotyp eine Rolle für das SCD-Risiko spielt, wobei Patienten mit krankheitsverursachenden Varianten in PLN, DSP, LMNA, FLNC, TMEM43 und RBM20 unabhängig von der LVEF eine signifikant höhere Rate an schweren Herzrhythmusstörungen aufweisen als Patienten mit anderen Ursachen für DCM unabhängig von der LVEF (3). Somit sollte beim Nachweis solcher Varianten eine ICD-Implantation in einem früheren Stadium evaluiert werden.
Die zunehmende Durchführbarkeit von Gentests in Verbindung mit der Verfügbarkeit diagnostischer Untersuchungen für DCM-Patienten könnte es in Zukunft ermöglichen, detailliertere Risikostratifizierungsschemata zu definieren (16).
Management
Neben der möglichen ätiologiebasierten Therapie ist die Standardtherapie der symptomatischen DCM mit reduzierter LVEF gemäss den aktuellen ESC-Leitlinien für Herzinsuffizienz wie folgt: Angiotensin-Converting-Enzym-Inhibitoren (ACE-I) oder Angiotensin-Rezeptor-Blocker (ARB), in Verbindung mit β-Blockern, Mineralcorticoid-Antagonisten (MRA) und in ausgewählten Fällen Vasodilatatoren (19). In letzter Zeit wurden die Angiotensin-Rezeptor-Neprilysin Inhibitoren (ARNI) und Ivabradin in die Liste der wirksamen Behandlungen für diejenigen, die nicht auf eine optimale medizinische Therapie ansprechen, hinzugefügt. Bei anhaltenden Symptomen (NYHA ≥ II) trotz optimaler Therapie, linksventrikulärer systolischen Dysfunktion (LVEF ≤ 35 %) und ventrikulären Asynchronie (QRS-Dauer ≥ 130 ms) ist eine kardiale Resynchronisationstherapie (CRT) indiziert (19).
Arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC)
Definition
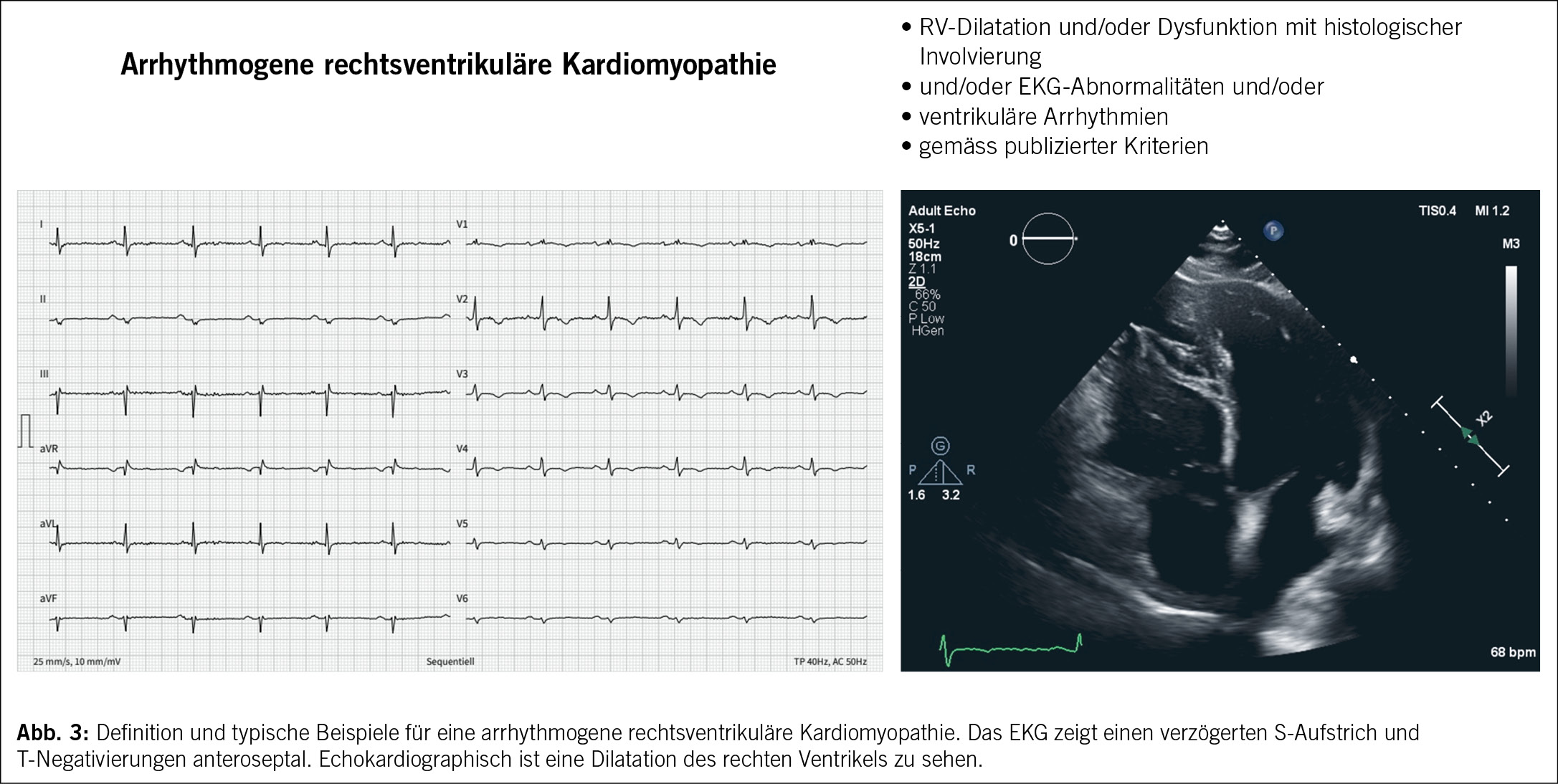
Die arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC) wird als eine dominant genetisch determinierte Herzmuskelerkrankung definiert. Die Pathologie ist durch einen fettig-fibrösen Ersatz des rechtsventrikulären (und linksventrikulären) Myokards charakterisiert (20). Klinisch kommt es zu ausgeprägten ventrikulären Arrhythmien und eine Beeinträchtigung der systolischen Funktion des Ventrikels (20). (Abb. 3)
Nomenklatur
Bezüglich der korrekten Nomenklatur für die Erkrankung besteht eine jahrelange Diskussion. Aktuell werden dominant zwei Termini parallel verwendet: (i) die «arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC)», welcher die Dominanz der rechtsventriuklären Veränderungen betont und in den aktuellen ESC-Richtlinien verwendet wird (3). Linksventrikuläre Formen sollen somit als NDLVC diagnostiziert werden. (ii) Die «arrhythmogene Kardiomyopathie (ACM)», welche die biventrikulären, rechts- und linksventrikulären Formen unterstreicht und in den neuesten Diagnosekriterien verwendet wird (21). Wir haben uns für diesen Artikel für ARVC als Terminus entschieden.
Pathogenese
Die Pathogenese der ARVC beruht typischerweise auf Varianten in Genen, welche hauptsächlich für Proteine des kardialen Desmosoms, aber auch der Kernhülle, des Natriumkanals oder des Sarkomers kodieren (20). Die Desmosomen sind für die Zell-Zell-Adhäsion verantwortlich und wichtige Mediatoren der intra- und interzelluläre Signalwege. Der Verlust der Adhäsion zwischen den Myozyten führt dazu, dass sie sich voneinander lösen und es dann allmählich zum Absterben der Herzmuskelzellen kommt. Somit wird dies durch fibröses oder faserig-fettiges Gewebe ersetzt, welches oft durch Entzündungen vermittelt wird. Dieses gilt als wesentlicher Mechanismus der Kardiomyopathie (22, 23).
Der Trigger ist die körperliche Belastung, die diesen Adhäsionsdefekt verschlimmert, wobei der rechte Ventrikel stärker betroffen ist, da seine Wand dünner ist als die des linken Ventrikels (1). Die Myokardatrophie ist ein fortlaufender Prozess, welcher vom Epikard beginnt und sich in Richtung Endokard ausbreitet und schliesslich transmural wird. Somit kommt es zu einer progressiven Ausdünnung der Wände und schliesslich zur Aneurysmabildung (1). So wird bei Patienten, die kompetitiven Sport betreiben, ein früheres Einsetzen von Symptomen sowie ein höheres Risiko für ventrikuläre Arrhythmien beobachtet (24). In einigen Fällen verläuft dieser Prozess jedoch nicht kontinuierlich, sondern in Form von periodischen akuten Schüben einer ansonsten stabilen Erkrankung, die eine Myokarditis imitieren, was eine wichtige Rolle bei der Pathogenese des Phänotyps spielen könnte (1, 20). So zeigen eine Grosszahl der autoptisch untersuchten Herzen mit ARVC ein entzündliches Infiltrat (20).
Klinik
Die Symptomatik der ARVC kann stark variieren, und zwar von asymptomatischen Familienmitgliedern mit verborgenen strukturellen Anomalien und ohne Arrhythmien bis hin zu symptomatischen Patienten, die einen plötzlichen Herztod erleiden oder die sich aufgrund einer therapierefraktären Herzinsuffizienz einer Herztransplantation unterziehen müssen (1, 25). Die häufigste klinische Präsentation ist zur Abklärung unspezifischer kardialer Symptome wie z.B. Palpitationen, Dyspnoe oder Synkopen, oder aber, die Patienten werden wegen Arrhythmien oder zum Familienscreening vorstellig (26). Leider kommt es immer wieder vor, dass bislang komplett unauffällige Patienten sich erstmals mit einem plötzlichen Herztod manifestieren (20).
Diagnose
Die Diagnose erfolgt durch verschiedene Modalitäten und wurde unter anderem durch die 2010 Task Force Kriterien (27) definiert, welche zuletzt durch die Padua-Kriterien ersetzt wurden, die auf verschiedenen Aspekten wie strukturellen Veränderungen, Depolarisations-/Repolarisationsstörungen, Arrhythmien und Familienanamnese beruhen (27). Um eine ARVC diagnostizieren zu können, müssen mindestens zwei unterschiedliche diagnostische Modalitäten eingesetzt werden. Dabei ist zu berücksichtigen, dass elektrische Veränderungen vor sichtbaren strukturellen Modifikationen auftreten können (28). Um die sichere Diagnose einer ARVC stellen zu können, müssen zwei Hauptkriterien oder ein Hauptkriterium und zwei sekundäre Kriterien oder vier sekundäre Kriterien aus verschiedenen Kategorien erfüllt werden. Für eine Borderline-Diagnose genügen ein Hauptkriterium und ein Nebenkriterium oder drei Kriterien aus verschiedenen Kategorien und für eine mögliche Diagnose ein Hauptkriterium oder zwei Nebenkriterien aus verschiedenen Kategorien (27).
Natürlicher Verlauf der Erkrankung
Der Verlauf der ARVC ist, wie bei allen Kardiomyopathien, höchst variabel und reicht von relativ oligosymptomatischen Patienten zu Patienten mit anhaltenden ventrikulären Arrhythmien, plötzlichem Herztod und Herzinsuffizienz. (25, 26, 29). Hier ist speziell zu erwähnen, dass der plötzliche Herztod als Endpunkt in allen Beobachtungsstudien relativ selten auftrat, obwohl durchschnittlich ein Risiko für anhaltende ventrikulären Arrhythmien von 3.7 – 10.6%/Jahr angegeben wird. (3) Dies hängt speziell damit zusammen, dass ARVC-Patienten öfters an anhaltenden ventrikulären Arrhythmien leiden, welche hämodynamisch gut toleriert werden und entsprechend nicht zu einem plötzlichen Herztod führen (3, 25).
Risikostratifizierung
Die grösste Herausforderung bei der Behandlung der ARVC ist nach wie vor die Risikostratifizierung für den plötzlichen Herztod und somit die Indikationsstellung für die ICD-Implantation. Das Hauptproblem bei der Indikationsstellung für einen ICD ist zwischen Komplikationen, fehlendem Nutzen und geretteten Leben abzuwägen. Basierend auf Daten zur jährlichen Mortalität in Verbindung mit Risikofaktoren teilt der Konsensalgorithmus der International Task Force die Patienten in drei verschiedene Risikokategorien ein (29). Das geschätzte Risiko für schwere arrhythmische Ereignisse liegt in der Hochrisikokategorie bei > 10 %/Jahr, in der Zwischenrisikokategorie zwischen 1 und 10 %/Jahr und in der Niedrigrisikokategorie bei < 1 %/Jahr. Kürzlich wurde in einer retrospektiven Studie mit ARVC-Patienten aus fünf Registern ein neues Modell zur Vorhersage des 5-Jahres-Risikos für ventrikuläre Arrhythmien bei ARVC vorgestellt, das vielversprechende, wenn auch nicht unumstrittene Ergebnisse liefert (31). Dieses Prognosemodell (ARVC Risk Calculator, www.arvcrisk.com) umfasst sieben Variablen: Geschlecht, Alter, kürzlich aufgetretene kardiale Synkopen (<6 Monate), nicht anhaltende ventrikuläre Tachykardien, 24-Stunden-VES (ventrikulkäre Extrasystolen)-Anzahl, Anzahl der Ableitungen mit T-Wellen-Inversion in den präkordialen und inferioren Ableitungen und rechtsventrikuläre Ejektionsfraktion (31). In der Originalpublikation wurde dieser Score jedoch nicht extern validiert. Mehrere Studien untersuchten die Leistungsfähigkeit des ARVC-Risikorechners in externen, unabhängigen Kohorten und zeigten, dass das Modell grundsätzlich Patienten mit einem erhöhten Risiko für ventrikuläre Arrhythmien gut identifiziert (32, 33). Es ist jedoch zu bemerken, dass der Rechner das Risiko für anhaltende ventrikuläre Arrhythmien – und nicht SCD – prognostiziert und dass das Risiko insgesamt, speziell aber bei Patienten mit niedrigem bis mittlerem Risiko, tendenziell überschätzt und die Aussagekraft bei Patienten Varianten in anderen Genen als PKP2 oder fehlenden pathogenen Varianten und nicht rechtsdominanten Formen der Erkrankung deutlich eingeschränkt ist (23, 33). Bislang wurde kein Schwellenwert validiert. Die ICD-Indikation sollte somit eine gemeinsame Entscheidung bleiben, die nicht nur auf dem Ergebnis des Scores, sondern auch auf der Gesamtbeurteilung des Patienten beruht (23).
Management
Das wichtigste klinische Ziel bei der Behandlung von ARVC ist die Verhinderung von SCD. Die derzeitigen therapeutischen Optionen umfassen Lebensstiländerungen, pharmakologische Optionen (β-Blocker und Antiarrhythmika), Katheterablation, ICD-Implantation und Herztransplantation (1).
Da körperliche Aktivität ein wichtiger Faktor ist, der die phänotypische Manifestation der Erkrankung favorisieren und damit lebensbedrohliche ventrikuläre Arrhythmien begünstigen kann, empfehlen die aktuellen Leitlinien die Abstinenz von Leistungssport für alle betroffenen Patienten und für Patienten mit einem hohen Risiko aufgrund einer pathogenen Variante (3). Leichte bis mittelschwere körperliche Aktivität können je nach Situation in einem gewissen Ausmass empfohlen werden – je intensiver die Aktivität, desto seltener sollte sie ausgeübt werden, im Sinne einer partizipativen Entscheidungsfindung (34).
Die Indikation für den Einsatz von β-Blockern bei ARVC beruht auf ihrer nachgewiesenen Wirksamkeit zur Reduktion der Inzidenz schwerer ventrikulärer Arrhythmien und in der Behandlung der Herzinsuffizienz (35). Daher sollte eine β-Blocker-Therapie bei allen Patienten mit eindeutiger Diagnose einer ARVC, speziell mit ventrikulären Arrhythmien (auch VES) empfohlen werden (3). Die vorliegende Evidenz zeigt, dass die Therapie mit Antiarrhythmika (Amiodaron und Sotalol) keinen ausreichenden Schutz vor plötzlichem Herztod bietet (35). Ferner sollte eine antiarrhythmische Therapie in Betracht gezogen werden, um die Arrhythmielast bei symptomatischen Patienten mit häufigen vorzeitigen ventrikulären Kontraktionen und nicht anhaltenden ventrikulären Tachykardien zu reduzieren (29).
Patienten mit ARVC, die eine rechts- oder biventrikuläre Herzinsuffizienz entwickeln – laut Literatur bis zu 13 % –, werden gemäss den ESC-Leitlinien 2016 mit Angiotensin-Converting-Enzym-Hemmern und Diuretika behandelt. Eine Herztransplantation kann die letzte Option bei schwerer Herzinsuffizienz sein, die auf die Standardtherapie nicht anspricht, oder bei unkontrollierbaren ventrikulären Tachyarrhythmien (25, 29). Zusätzlich sollte bei Patienten mit wiederholten appropriaten ICD-Therapien trotz β-Blockern eine Katheterablation der Kammertachykardien in Erwägung gezogen werden (3).
Nicht-dilatierte linksventrikuläre Kardiomyopathie (NDLVC)
Definition
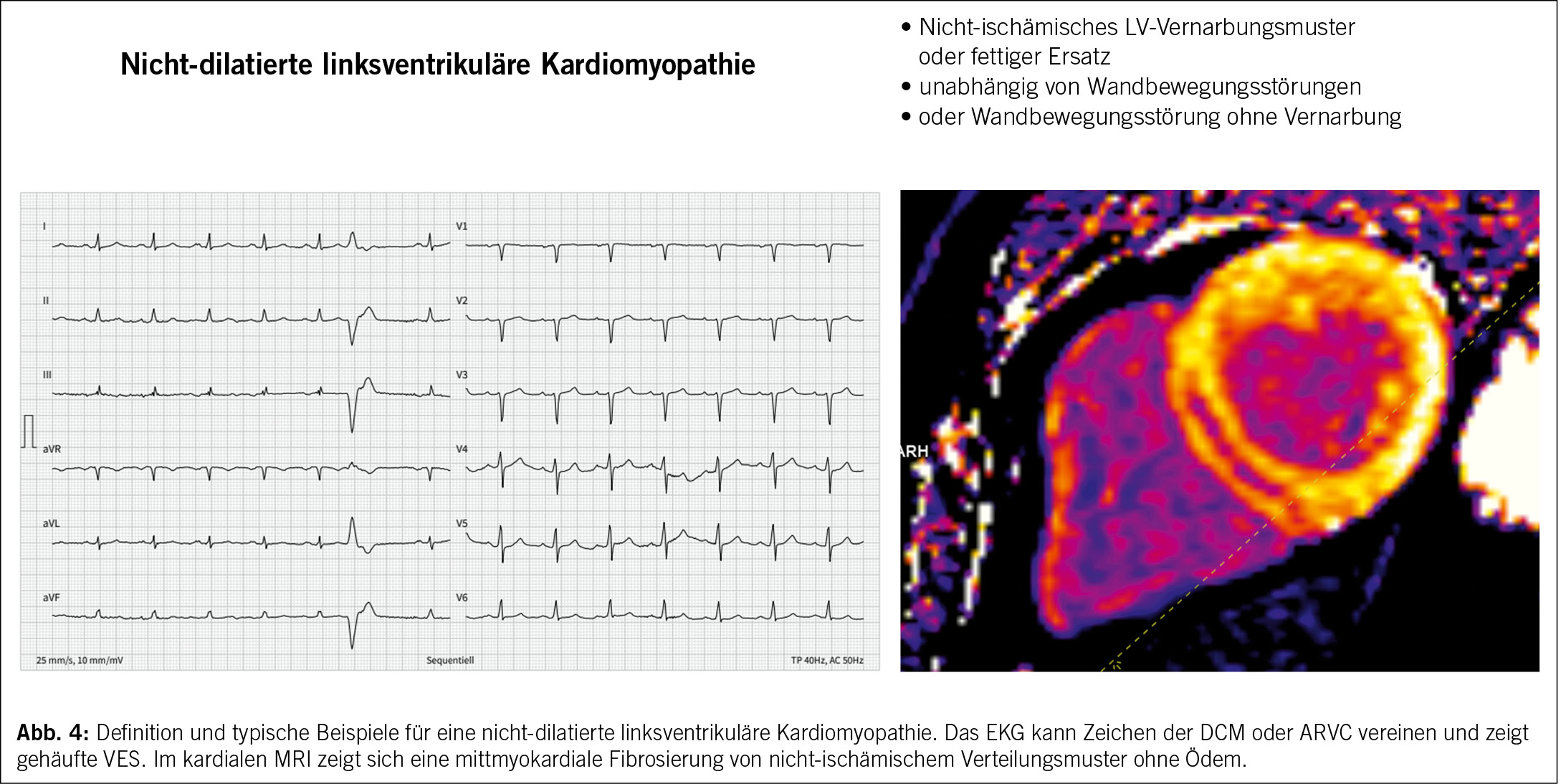
Die bisherige Definition von DCM und ARVC hat zur Folge, dass frühe und intermediäre Formen genetischer und erworbener Erkrankungen nicht genügend abgebildet wurden und somit das Risiko besteht, dass sie übersehen werden. Unter dieser Form werden Entitäten subsummiert, die früher z.B. als DCM ohne Dilatation, arrhythmogene linksventrikuläre Kardiomyopathie oder arrhythmogene DCM bezeichnet wurden. Bei dieser Form soll ein spezielles Augenmerk auf eine multiparametrische Abklärung, insbesondere auch die genetische Abklärung, geworfen werden, da die spezifische Ätiologie die klinische Behandlung beeinflusst. (Abb. 4)
Abkürzungen
AAD Antiarrhythmische Medikamente
ACE-I Angiotensin-Converting-Enzym-Inhibitoren
ACM Arrhythmogene Kardiomyopathie
ARB Angiotensin-Rezeptor-Blocker
ARNI Angiotensin-Rezeptor-Neprilysin Inhibitoren
ARVC Arrhythmogenic right ventricular cardiomyopathy
CRT kardiale Resynchronisationstherapie
DCM Dilatative Kardiomyopathie
ESC Europäische Gesellschaft für Kardiologie
HCM Hypertrophe Kardiomyopathie
ICD Implantable Cardioverter Defibrillator
LVEF linksventrikuläre Ejektionsfraktion
LVOT Linksventrikulärer Ausflusstrakt
MRA Mineralcorticoid-Antagonisten
MYBPC3 Myosin-bindendes Protein C
MYH7 schwere β-Myosinkette 7
NDLVC Nicht-dilatierte linksventrikuläre Kardiomyopathie
NYHA New York Heart Association
SCD Plötzlicher Herztod
VA Ventrikuläre Arrhythmien
VES Ventrikuläre Extrasystolen
Copyright
Aerzteverlag medinfo AG
Medizinische Poliklinik
Universitätsspital Basel
Petersgraben 4
4031 Basel
Medizinische Poliklinik
Universitätsspital Basel
Petersgraben 4
4031 Basel
PD Dr. A. Vischer hat Honorare von Bristol Myers Squibb, Amarin, Servier, Medtronic, Vifor und NovoNordisk erhalten.
1. Schulz LP, Vischer AS. Cardiomyopathies in the Clinical Practice – an Overview. Praxis (Bern 1994). 2022;111(11):623-31.
2. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270-6.
3. Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44(37):3503-626.
4. Miles C, Fanton Z, Tome M, Behr ER. Inherited cardiomyopathies. Bmj. 2019;365:l1570.
5. Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733-79.
6. Zampieri M, Berteotti M, Ferrantini C, Tassetti L, Gabriele M, Tomberli B, et al. Pathophysiology and Treatment of Hypertrophic Cardiomyopathy: New Perspectives. Curr Heart Fail Rep. 2021;18(4):169-79.
7. Pantazis A, Vischer AS, Perez-Tome MC, Castelletti S. Diagnosis and management of hypertrophic cardiomyopathy. Echo Res Pract. 2015;2(1):R45-53.
8. O‘Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014;35(30):2010-20.
9. Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, Saberi S, Lakdawala NK, Wheeler MT, Owens A, Kubanek M, Wojakowski W, Jensen MK, Gimeno-Blanes J, Afshar K, Myers J, Hegde SM, Solomon SD, Sehnert AJ, Zhang D, Li W, Bhattacharya M, Edelberg JM, Waldman CB, Lester SJ, Wang A, Ho CY, Jacoby D; EXPLORER-HCM study investigators. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020 Sep 12;396(10253):759-769. doi: 10.1016/S0140-6736(20)31792-X. Epub 2020 Aug 29. Erratum in: Lancet. 2020 Sep 12;396(10253):758. doi: 10.1016/S0140-6736(20)31872-9.
10. Ommen SR, Ho CY, Asif IM, Balaji S, Burke MA, Day SM, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2024;149(23):e1239-e311.
11. Guttmann OP, Rahman MS, O‘Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart. 2014;100(6):465-72.
12. Sorajja P, Ommen SR, Nishimura RA, Gersh BJ, Berger PB, Tajik AJ. Adverse prognosis of patients with hypertrophic cardiomyopathy who have epicardial coronary artery disease. Circulation. 2003;108(19):2342-8.
13. Merlo M, Cannatà A, Gobbo M, Stolfo D, Elliott PM, Sinagra G. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20(2):228-39.
14. Ware JS, Amor-Salamanca A, Tayal U, Govind R, Serrano I, Salazar-Mendiguchía J, et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J Am Coll Cardiol. 2018;71(20):2293-302.
15. Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J Am Coll Cardiol. 2019;74(11):1480-90.
16. Schultheiss HP, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE, et al. Dilated cardiomyopathy. Nat Rev Dis Primers. 2019;5(1):32.
17. Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850-8.
18. Paldino A, De Angelis G, Merlo M, Gigli M, Dal Ferro M, Severini GM, et al. Genetics of Dilated Cardiomyopathy: Clinical Implications. Curr Cardiol Rep. 2018;20(10):83.
19. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. 2021;42(36):3599-726.
20. Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017;121(7):784-802.
21. Corrado D, Anastasakis A, Basso C, Bauce B, Blomström-Lundqvist C, Bucciarelli-Ducci C, et al. Proposed diagnostic criteria for arrhythmogenic cardiomyopathy: European Task Force consensus report. Int J Cardiol. 2024;395:131447.
22. Lin YN, Ibrahim A, Marbán E, Cingolani E. Pathogenesis of arrhythmogenic cardiomyopathy: role of inflammation. Basic Res Cardiol. 2021;116(1):39.
23. Gandjbakhch E, Redheuil A, Pousset F, Charron P, Frank R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72(7):784-804.
24. Ruwald AC, Marcus F, Estes NA, 3rd, Link M, McNitt S, Polonsky B, et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36(27):1735-43.
25. Vischer AS, Castelletti S, Syrris P, McKenna WJ, Pantazis A. Heart failure in patients with arrhythmogenic right ventricular cardiomyopathy: Genetic characteristics. Int J Cardiol. 2019;286:99-103.
26. Vischer AS, Castelletti S, Syrris P, Bastiaenen R, Miles C, Akdis D, et al. Risk score for the exclusion of arrhythmic events in arrhythmogenic right ventricular cardiomyopathy at first presentation. Int J Cardiol. 2019;290:100-5.
27. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31(7):806-14.
28. Akdis D, Brunckhorst C, Duru F, Saguner AM. Arrhythmogenic Cardiomyopathy: Electrical and Structural Phenotypes. Arrhythm Electrophysiol Rev. 2016;5(2):90-101.
29. Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet. 2015;8(3):437-46.
30. Corrado D, Wichter T, Link MS, Hauer R, Marchlinski F, Anastasakis A, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Eur Heart J. 2015;36(46):3227-37.
31. Cadrin-Tourigny J, Bosman LP, Nozza A, Wang W, Tadros R, Bhonsale A, et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2019;40(23):1850-8.
32. Jordà P, Bosman LP, Gasperetti A, Mazzanti A, Gourraud JB, Davies B, et al. Arrhythmic risk prediction in arrhythmogenic right ventricular cardiomyopathy: external validation of the arrhythmogenic right ventricular cardiomyopathy risk calculator. Eur Heart J. 2022;43(32):3041-52.
33. Protonotarios A, Bariani R, Cappelletto C, Pavlou M, García-García A, Cipriani A, et al. Importance of genotype for risk stratification in arrhythmogenic right ventricular cardiomyopathy using the 2019 ARVC risk calculator. Eur Heart J. 2022;43(32):3053-67.
34. Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm. 2019;16(11):e373-e407.
35. Cappelletto C, Gregorio C, Barbati G, Romani S, De Luca A, Merlo M, et al. Antiarrhythmic therapy and risk of cumulative ventricular arrhythmias in arrhythmogenic right ventricle cardiomyopathy. Int J Cardiol. 2021;334:58-64.


info@herz+gefäss
- Vol. 15
- Ausgabe 1
- März 2025