Morbus Fabry ist eine seltene X-chromosomale lysosomale Speichererkrankung, bei der es durch Enzymmangel (α-Galacto-sidase A) zur Glykosphingolipid-Ablagerung in verschiedenen Organen, vor allem dem Herzen und den Nieren, kommt und die mittels Enzymersatztherapie behandelt werden kann. Typisch ist eine konzentrische linksventrikuläre Hypertrophie sowie Myokardfibrose, die zur Herzinsuffizienz und Arrhythmien führen kann. Letztere sind die wichtigsten Todesursachen.
Morbus Fabry ist eine X-chromosomal vererbte lysosomale Speichererkrankung mit einer weltweit geschätzten Inzidenz von 1:40 000 (1), bei der es durch Mangel des Enzyms α-Galactosidase A zur Ablagerung von Glykosphingolipiden, insbesondere Globotriaosylceramid (GB3), in verschiedenen Organen kommt (2) und die in einer Multisystemerkrankung mit vordergründig kardialer und renaler Beteiligung resultiert. Weitere Organe, die betroffen sein können, sind die Haut, die Augen und das Nervensystem. Die typische Herzmanifestation ist eine konzentrische linksventrikuläre Hypertrophie (LVH). Bei idiopathischen Formen der LVH wird mit 1-6% auch nicht selten als Ursache ein Morbus Fabry gefunden (3-6). Die Haupttodesursache bei dieser Erkrankung sind kardiovaskuläre Ereignisse, primär Arrhythmien (7). Leider werden Fabry-Patienten oft immer noch erst lange nach Symptombeginn, im Durchschnitt 13.7 Jahre bei Männern und 16.3 Jahren bei Frauen, diagnostiziert (8). Während bei Männern die Diagnose mittels Enzymaktivitätsbestimmung in den Leukozyten gestellt wird, ist bei Frauen ein Gentest diagnostisch, da sie trotz einer Erkrankung eine normale Enzymaktivität haben können.
Klinik
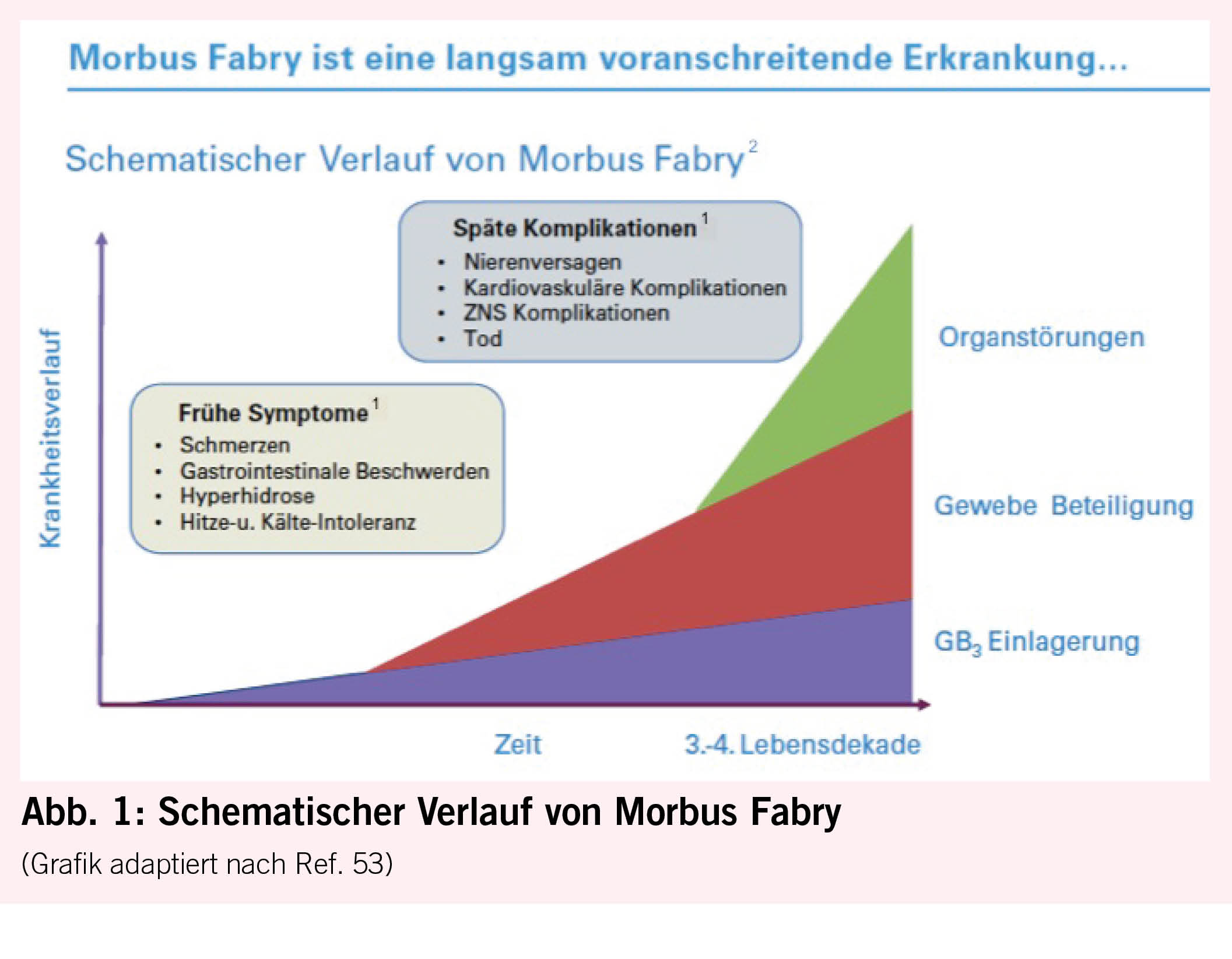
Bei Morbus Fabry unterscheidet man klassische von atypischen Krankheitsmanifestationen. Der klassische Phänotyp tritt bei homozygoten Männern auf, die keine Enzymaktivität aufweisen und daher oft schon früh in der Kindheit oder Jugend unter febrilen Schmerzkrisen, Akroparästhesien, Hypohidrose, Angiokeratomen und gastrointestinalen Beschwerden, vor allem Durchfall, leiden. Durch progrediente GB3-Ablagerungen kann es dann im Erwachsenenalter zu schweren Schädigungen des Herzens (Herzrhythmusstörungen und Herzinsuffizienz), der Nieren (bis hin zur Dialyse oder Nierentransplantation) und des Gehirns (zerebrovaskuläre Ereignisse) kommen (Abb. 1). Während heterozygote Frauen früher oft nur als asymptomatische Konduktorinnen galten, ist heute bekannt, dass sie genauso auch einen Vollphänotyp entwickeln können, der eher später im Leben auftritt als bei Männern. Die atypische Form ist durch eine reduzierte, aber noch nachweisbare Enzymaktivität gekennzeichnet, die mit einem späteren Symptombeginn mit Oligosymptomatik und isolierter Herz- oder Nierenbeteiligung einhergeht. Die letztere Form wird oft bei der Abklärung einer idiopathischen LVH (6, 9) oder Niereninsuffizienz entdeckt.
Fabry-Kardiomyopathie
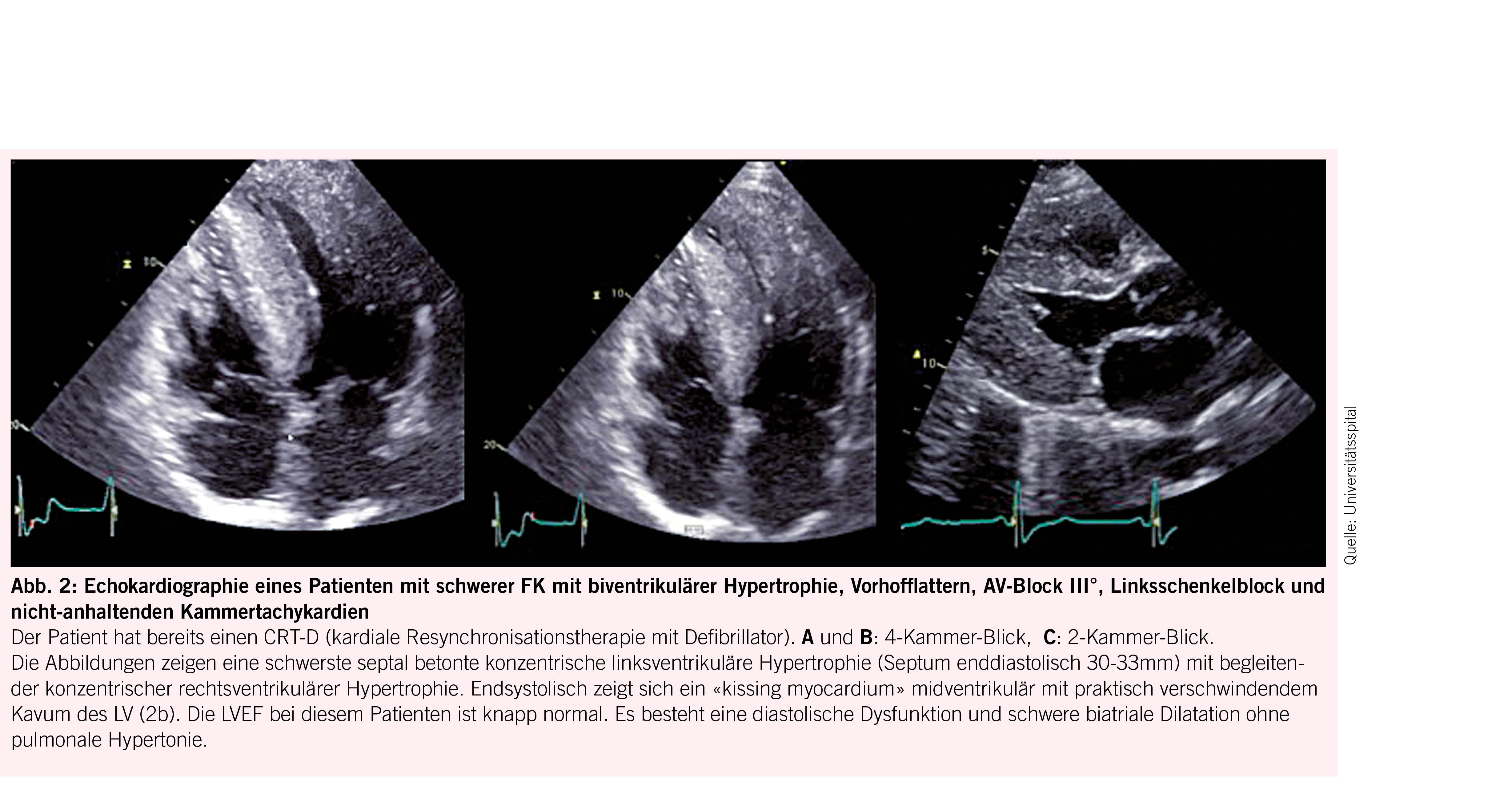
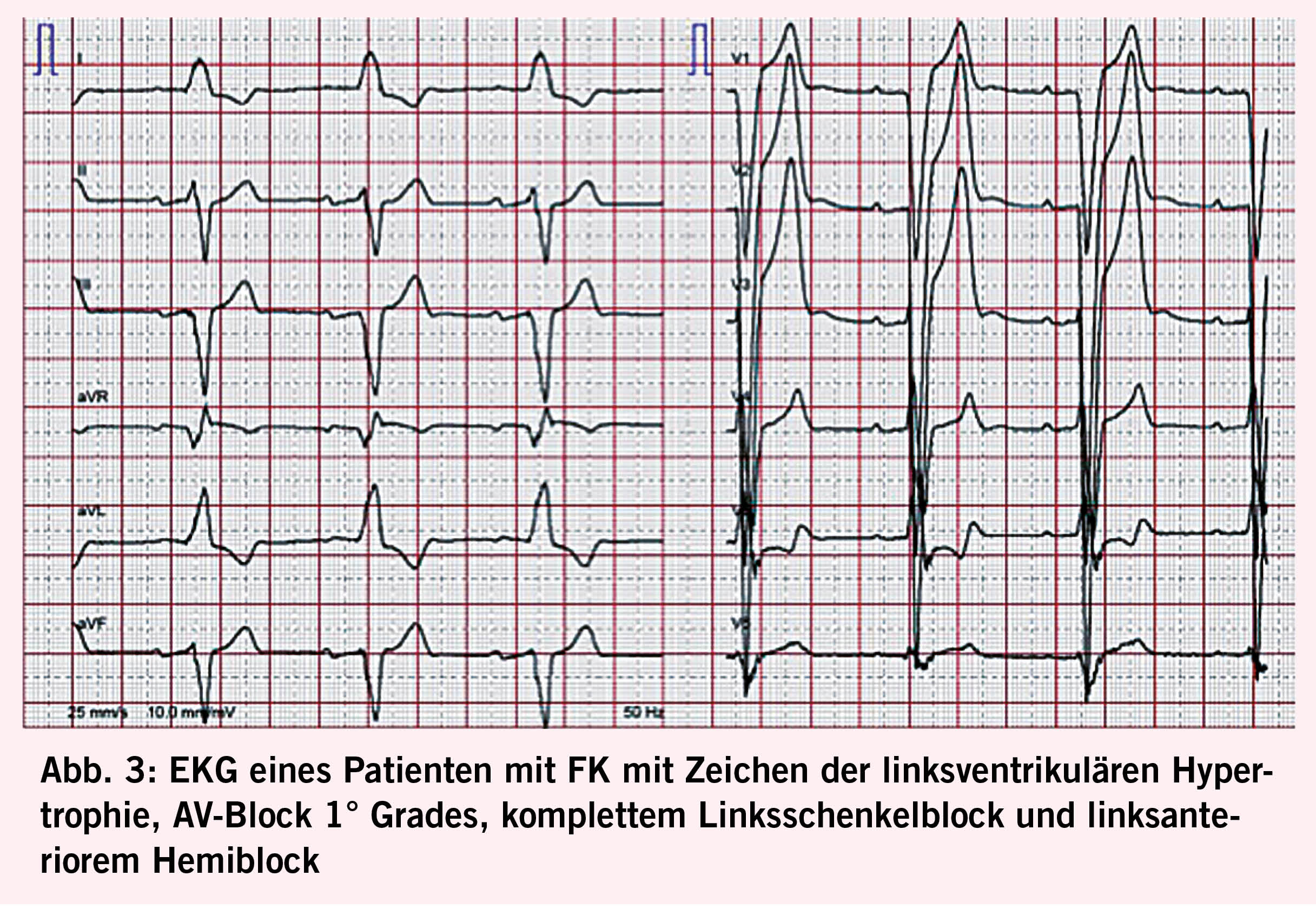
Das Herz ist mit 68% das Organ mit der häufigsten intrazellulären GB3-Akkumulation (10,11), die in den Myozyten, Herzklappen und Gefässendothelien stattfindet und neben der Infiltration auch einen inflammatorischen und oxidativen Stress verursacht (12). Als Folge leiden über die Hälfte der männlichen Fabry-Patienten und 18- 28% der Frauen mit Morbus Fabry (13, 14) unter einer konzentrischen LVH (siehe Abb. 2) mit einer typischerweise früh einsetzenden diastolischen Dysfunktion (15,16) und im Verlauf restriktivem Füllungsmuster (17, 18). Die LVH bei Fabry unterscheidet sich in der Regel durch das Fehlen einer Ausflussobstruktion von einer idiopathischen hypertrophischen Kardiomyopathie (4) und kann auch mit einer rechtsventrikulären Hypertrophie und Dysfunktion einhergehen (19). Während bei Männern erste kardiale Beschwerden mit durchschnittlich 33 Jahren einsetzen, wurde bei Frauen ein späterer Beginn um das 40. Lebensjahr beobachtet (7). Typischerweise kommt es im Verlauf zu einer Herzinsuffizienz mit erhaltener Ejektionsfraktion (7). Aufgrund der Endothelablagerungen kann es zudem oft auch zu einer endothelialen Dysfunktion (20, 21) kommen. Möglich ist auch eine mikrovaskuläre Dysfunktion, die Angina pectoris verursacht (22). Die fortgeschrittene Fabry-Kardiomyopathie (FK) ist durch eine fibrotische Umwandlung des linken Ventrikels, die typischerweise infero-baso-lateral beginnt und nach transmural fortschreitet, gekennzeichnet (23). Diese Fi-broseareale können Rhythmusstörungen (siehe Abb. 3), darunter Sinusbradykardien, höhergradige AV-Blockierungen, und Kammertachykardien hervorrufen und infolge zum plötzlichen Herztod führen (24-26). Auch Vorhofflimmern ist bei Fabry-Patienten häufig zu finden, wobei die Inzidenz vier Mal so hoch ist wie in der Normalbevölkerung, bei über 50-jährigen Patienten sogar zwölffach erhöht (25). Bei der fortgeschrittenen FK kommt es gelegentlich auch zur Herzklappenschädigung, insbesondere zur Insuffizienz der Aorten-, Mitral- oder Trikuspidalklappe (24) sowie Erweiterung der Aortenwurzel (27). Eine weitere typische Veränderung ist ein prominenter Papillarmuskel (28) (Abb. 2).
Untersuchungen
Die transthorakale Echokardiografie ist die primäre und am leichtesten verfügbare Methode zum Screening und zur Verlaufsbeurteilung bei FK (Abb. 2). Sie ermöglicht es jedoch nicht, eine Fabrykardiomyopathie von anderen Hypertrophien abzugrenzen und eine Herzbeteiligung vor Beginn der LVH zu erkennen. Letzteres ist gerade bei Frauen relevant, da diese eine Myokardfibrose oft noch vor den Zeichen einer LVH entwickeln (29). Insgesamt haben mittlerweile 60% der Patienten bei der Diagnosestellung noch keine nachweisbare LVH (30). Durch den Einsatz neuerer Techniken (z.B. Strain Analyse und 2-D Speckle Tracking) können jedoch myokardiale regionale Unterschiede sowie eine beginnende diastolische Dysfunktion vor der LVH detektiert werden (14, 31). Den Goldstandard für die Beurteilung struktureller Veränderungen bei Morbus Fabry sowie die Feststellung von Myokardfibrosen stellt die kardiale Magnetresonanztomographie (CMR) dar. Sie ermöglicht 1. mittels «Late Gadolinium Enhancement» eine frühzeitige Erfassung von Fibrosearealen (16, 32), was von grosser prognostischer Bedeutung ist, da deren Vorhandensein und Ausmass mit dem Risiko des Auftretens potentiell lebensbedrohlicher Rhythmusstörungen korreliert (33) und bei der Indikationsstellung zur ICD-Implantation helfen kann (34), 2. mittels nativem T1 Mapping mit hoher Spezifität und Sensitivität eine FK von anderen Kardiomyopathien zu differenzieren (sehr niedriger T1 Map Wert) (19, 32, 35), und 3. Verlaufskontrollen des Remodelings unter ERT durchzuführen (10, 36). Bei Kontraindikationen für eine CMR kann die Frage nach einer mikrovaskulären Dysfunktion auch alternativ mittels kardialer Positronen-Emissions-Tomographie untersucht werden (37). Im Ruhe-Elektrokardiogramm (EKG, siehe Abb. 3) lassen sich oft charakteristische Veränderungen wie ein positiver Sokolow-Lyon-Index und präkordiale T-Wellen-Negativierungen sowie gelegentlich auch eine PQ-Zeit-Verkürzung und QTc-Zeit-Verlängerung feststellen (38). Bei allen Patienten mit Symptomen sowie bei bekannter Myokardfibrose sollte regelmässig ein Holter-EKG durchgeführt werden. Bei unauffälligem Holter-EKG und persistierendem Verdacht auf eine seltener auftretende HRST sollte die Implantation eines Event Recorders erwogen werden (39). Als Verlaufsparameter, der mit dem Fibrosegrad und dem Schweregrad der FK korreliert, kann hs-Troponin eingesetzt werden (40).
Therapie
Seit 2001 können Fabrypatienten mittels intravenöser Enzymersatztherapie (ERT) behandelt werden, die in der Regel in 14-tägigen Abständen durchgeführt wird. Die ERT ist die bisher einzige etablierte kausale und potentiell krankheitsstabilisierende Therapie (41, 42), durch die insbesondere auch das kardiale Outcome verbessert werden kann (36). Bei einigen bestimmten Mutationen ist neu seit 2016 eine orale Chaperone-Therapie (Migalastat) verfügbar (43). Ein früher Beginn der Behandlung, am besten noch vor dem Auftreten einer Myokardfibrose, ist wichtig, um den besten Therapieeffekt zu erreichen (44, 45). So ist im frühen Krankheitsstadium teilweise sogar eine Reversibilität der LVH möglich (36). Medikamentös kann durch den Einsatz von Angiotensin-Converting-Enzyme (ACE)-Hemmern oder Angiotensin-Rezeptor-Blockern eine Progression der LVH verlangsamt (46) und die Nierenfunktion stabilisiert werden (47). Ausserdem ist im Allgemeinen bei Fabrypatienten eine optimale Blutdruckeinstellung von besonderer Bedeutung (48). Bei tachykarden HRST und zur Vorbeugung ventrikulärer Rhythmusstörungen, bei Bradykardie natürlich mit Vorsicht, können Beta-Blocker eingesetzt werden. Bei bradykarden HRST sollte eine Herzschrittmacher-Implantation frühzeitig evaluiert werden (25). Aufgrund der Häufigkeit solcher Rhythmusstörungen benötigen im Krankheitsverlauf 10-20% der Patienten einen Herzschrittmacher (49). Bei fortgeschrittener FK mit LVH und Nachweis von Myokardfibrose sowie potentiell malignen ventrikulären HRST sollte eine primärprophylaktische ICD-Implantation erwogen werden (16, 50). Wegen des hohen Schlaganfallrisikos kann eine Dauertherapie mit Aspirin 100mg/Tag eingesetzt (49) und bei Vorhofflimmern unabhängig vom CHA2DS2-VASc Score antikoaguliert werden (10). Bei terminaler Herzinsuffizienz stellt eine Herztransplantation die Ultima Ratio dar. Ein relevantes Rezidiv auf dem Transplantat ist bei behandelten Patienten weder für das Herz (51) noch für die Nieren beschrieben worden (52).
Oberärztin Klinik für Endokrinologie
Rare Diseases
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
Albina.Nowak@usz.ch
Universitätsklinik für Kardiologie
Schweizer Herz- und Gefässzentrum Bern
Inselspital
Universitätsspital Bern
3010 Bern
Sara.Ersoezlue@insel.ch
Die Autorinnen haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel.
Literatur
1. Duro G, Musumeci MB, Colomba P, Zizzo C, Albeggiani G, Mastromarino V, Volpe M, Autore C: Novel alpha-galactosidase A mutation in patients with severe cardiac manifestations of Fabry disease. Gene, 535: 365-369, 2014
2. Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L: Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med, 276: 1163-1167, 1967
3. Palecek T, Honzikova J, Poupetova H, Vlaskova H, Kuchynka P, Golan L, Magage S, Linhart A: Prevalence of Fabry disease in male patients with unexplained left ventricular hypertrophy in primary cardiology practice: prospective Fabry cardiomyopathy screening study (FACSS). J Inherit Metab Dis, 37: 455-460, 2014
4. Nakao S, Takenaka T, Maeda M, Kodama C, Tanaka A, Tahara M, Yoshida A, Kuriyama M, Hayashibe H, Sakuraba H, et al.: An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med, 333: 288-293, 1995
5. Monserrat L, Gimeno-Blanes JR, Marin F, Hermida-Prieto M, Garcia-Honrubia A, Perez I, Fernandez X, de Nicolas R, de la Morena G, Paya E, Yague J, Egido J: Prevalence of fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol, 50: 2399-2403, 2007
6. Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ, Elliott PM: Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation, 105: 1407-1411, 2002
7. Linhart A, Kampmann C, Zamorano JL, Sunder-Plassmann G, Beck M, Mehta A, Elliott PM, European FOSI: Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J, 28: 1228-1235, 2007
8. Germain DP: Fabry disease. Orphanet J Rare Dis, 5: 30, 2010
9. Chimenti C, Pieroni M, Morgante E, Antuzzi D, Russo A, Russo MA, Maseri A, Frustaci A: Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation, 110: 1047-1053, 2004
10. Yogasundaram H, Kim D, Oudit O, Thompson RB, Weidemann F, Oudit GY: Clinical Features, Diagnosis, and Management of Patients With Anderson-Fabry Cardiomyopathy. Can J Cardiol, 33: 883-897, 2017
11. Favalli V, Disabella E, Molinaro M, Tagliani M, Scarabotto A, Serio A, Grasso M, Narula N, Giorgianni C, Caspani C, Concardi M, Agozzino M, Giordano C, Smirnova A, Kodama T, Giuliani L, Antoniazzi E, Borroni RG, Vassallo C, Mangione F, Scelsi L, Ghio S, Pellegrini C, Zedde M, Fancellu L, Sechi G, Ganau A, Piga S, Colucci A, Concolino D, Di Mascio MT, Toni D, Diomedi M, Rapezzi C, Biagini E, Marini M, Rasura M, Melis M, Nucera A, Guidetti D, Mancuso M, Scoditti U, Cassini P, Narula J, Tavazzi L, Arbustini E: Genetic Screening of Anderson-Fabry Disease in Probands Referred From Multispecialty Clinics. J Am Coll Cardiol, 68: 1037-1050, 2016
12. Linhart A: The heart in Fabry disease. In: Fabry Disease: Perspectives from 5 Years of FOS. edited by MEHTA, A., BECK, M., SUNDER-PLASSMANN, G., Oxford, 2006,
13. Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, Linhart A, Sunder-Plassmann G, Ries M, Beck M: Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest, 34: 236-242, 2004
14. Shanks M, Thompson RB, Paterson ID, Putko B, Khan A, Chan A, Becher H, Oudit GY: Systolic and diastolic function assessment in fabry disease patients using speckle-tracking imaging and comparison with conventional echocardiographic measurements. J Am Soc Echocardiogr, 26: 1407-1414, 2013
15. Barbey F, Brakch N, Linhart A, Rosenblatt-Velin N, Jeanrenaud X, Qanadli S, Steinmann B, Burnier M, Palecek T, Bultas J, Hayoz D: Cardiac and vascular hypertrophy in Fabry disease: evidence for a new mechanism independent of blood pressure and glycosphingolipid deposition. Arterioscler Thromb Vasc Biol, 26: 839-844, 2006
16. Weidemann F, Breunig F, Beer M, Sandstede J, Stork S, Voelker W, Ertl G, Knoll A, Wanner C, Strotmann JM: The variation of morphological and functional cardiac manifestation in Fabry disease: potential implications for the time course of the disease. Eur Heart J, 26: 1221-1227, 2005
17. Mehta A, Clarke JT, Giugliani R, Elliott P, Linhart A, Beck M, Sunder-Plassmann G, Investigators FOS: Natural course of Fabry disease: changing pattern of causes of death in FOS – Fabry Outcome Survey. J Med Genet, 46: 548-552, 2009
18. Garcia MJ: Constrictive Pericarditis Versus Restrictive Cardiomyopathy? J Am Coll Cardiol, 67: 2061-2076, 2016
19. Pagano JJ, Chow K, Khan A, Michelakis E, Paterson I, Oudit GY, Thompson RB: Reduced Right Ventricular Native Myocardial T1 in Anderson-Fabry Disease: Comparison to Pulmonary Hypertension and Healthy Controls. PLoS One, 11: e0157565, 2016
20. Rao DA, Lakdawala NK, Miller AL, Loscalzo J: Clinical problem-solving. In the thick of it. N Engl J Med, 368: 1732-1738, 2013
21. Arbustini E, Narula N, Dec GW, Reddy KS, Greenberg B, Kushwaha S, Marwick T, Pinney S, Bellazzi R, Favalli V, Kramer C, Roberts R, Zoghbi WA, Bonow R, Tavazzi L, Fuster V, Narula J: The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol, 62: 2046-2072, 2013
22. Chimenti C, Morgante E, Tanzilli G, Mangieri E, Critelli G, Gaudio C, Russo MA, Frustaci A: Angina in fabry disease reflects coronary small vessel disease. Circ Heart Fail, 1: 161-169, 2008
23. Moon JC, Sachdev B, Elkington AG, McKenna WJ, Mehta A, Pennell DJ, Leed PJ, Elliott PM: Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J, 24: 2151-2155, 2003
24. Weidemann F, Strotmann JM, Niemann M, Herrmann S, Wilke M, Beer M, Voelker W, Ertl G, Emmert A, Wanner C, Breunig F: Heart valve involvement in Fabry cardiomyopathy. Ultrasound Med Biol, 35: 730-735, 2009
25. Shah JS, Hughes DA, Sachdev B, Tome M, Ward D, Lee P, Mehta AB, Elliott PM: Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol, 96: 842-846, 2005
26. Baig S, Edward NC, Kotecha D, Liu B, Nordin S, Kozor R, Moon JC, Geberhiwot T, Steeds RP: Ventricular arrhythmia and sudden cardiac death in Fabry disease: a systematic review of risk factors in clinical practice. Europace, 2017
27. Fernandez LP, Lopez-Marquez A, Santisteban P: Thyroid transcription factors in development, differentiation and disease. Nat Rev Endocrinol, 11: 29-42, 2015
28. Kozor R, Callaghan F, Tchan M, Hamilton-Craig C, Figtree GA, Grieve SM: A disproportionate contribution of papillary muscles and trabeculations to total left ventricular mass makes choice of cardiovascular magnetic resonance analysis technique critical in Fabry disease. J Cardiovasc Magn Reson, 17: 22, 2015
29. Niemann M, Herrmann S, Hu K, Breunig F, Strotmann J, Beer M, Machann W, Voelker W, Ertl G, Wanner C, Weidemann F: Differences in Fabry cardiomyopathy between female and male patients: consequences for diagnostic assessment. JACC Cardiovasc Imaging, 4: 592-601, 2011
30. Linhart A, Palecek T, Bultas J, Ferguson JJ, Hrudova J, Karetova D, Zeman J, Ledvinova J, Poupetova H, Elleder M, Aschermann M: New insights in cardiac structural changes in patients with Fabry’s disease. Am Heart J, 139: 1101-1108, 2000
31. Soullier C, Obert P, Doucende G, Nottin S, Cade S, Perez-Martin A, Messner-Pellenc P, Schuster I: Exercise response in hypertrophic cardiomyopathy: blunted left ventricular deformational and twisting reserve with altered systolic-diastolic coupling. Circ Cardiovasc Imaging, 5: 324-332, 2012
32. Sado DM, White SK, Piechnik SK, Banypersad SM, Treibel T, Captur G, Fontana M, Maestrini V, Flett AS, Robson MD, Lachmann RH, Murphy E, Mehta A, Hughes D, Neubauer S, Elliott PM, Moon JC: Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging, 6: 392-398, 2013
33. Kramer J, Niemann M, Stork S, Frantz S, Beer M, Ertl G, Wanner C, Weidemann F: Relation of burden of myocardial fibrosis to malignant ventricular arrhythmias and outcomes in Fabry disease. Am J Cardiol, 114: 895-900, 2014
34. Sene T, Lidove O, Sebbah J, Darondel JM, Picard H, Aaron L, Fain O, Zenone T, Joly D, Charron P, Ziza JM: Cardiac device implantation in Fabry disease: A retrospective monocentric study. Medicine (Baltimore), 95: e4996, 2016
35. Thompson RB, Chow K, Khan A, Chan A, Shanks M, Paterson I, Oudit GY: T(1) mapping with cardiovascular MRI is highly sensitive for Fabry disease independent of hypertrophy and sex. Circ Cardiovasc Imaging, 6: 637-645, 2013
36. Weidemann F, Niemann M, Stork S, Breunig F, Beer M, Sommer C, Herrmann S, Ertl G, Wanner C: Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med, 274: 331-341, 2013
37. Tomberli B, Cecchi F, Sciagra R, Berti V, Lisi F, Torricelli F, Morrone A, Castelli G, Yacoub MH, Olivotto I: Coronary microvascular dysfunction is an early feature of cardiac involvement in patients with Anderson-Fabry disease. Eur J Heart Fail, 15: 1363-1373, 2013
38. Kramer J, Nordbeck P, Stork S, Ritter C, Ertl G, Wanner C, Weidemann F: Electrical Changes in Resting, Exercise, and Holter Electrocardiography in Fabry Cardiomyopathy. JIMD Rep, 2015
39. Weidemann F, Maier SK, Stork S, Brunner T, Liu D, Hu K, Seydelmann N, Schneider A, Becher J, Canan-Kuhl S, Blaschke D, Bijnens B, Ertl G, Wanner C, Nordbeck P: Usefulness of an Implantable Loop Recorder to Detect Clinically Relevant Arrhythmias in Patients With Advanced Fabry Cardiomyopathy. Am J Cardiol, 118: 264-274, 2016
40. Seydelmann N, Liu D, Kramer J, Drechsler C, Hu K, Nordbeck P, Schneider A, Stork S, Bijnens B, Ertl G, Wanner C, Weidemann F: High-Sensitivity Troponin: A Clinical Blood Biomarker for Staging Cardiomyopathy in Fabry Disease. J Am Heart Assoc, 5, 2016
41. Desnick RJ, Brady R, Barranger J, Collins AJ, Germain DP, Goldman M, Grabowski G, Packman S, Wilcox WR: Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med, 138: 338-346, 2003
42. Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, Ramaswami U, Rolfs A, Rivera A, Waldek S, Germain DP: Fabry disease: a review of current management strategies. QJM, 103: 641-659, 2010
43. Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, Feliciani C, Shankar SP, Ezgu F, Amartino H, Bratkovic D, Feldt-Rasmussen U, Nedd K, Sharaf El Din U, Lourenco CM, Banikazemi M, Charrow J, Dasouki M, Finegold D, Giraldo P, Goker-Alpan O, Longo N, Scott CR, Torra R, Tuffaha A, Jovanovic A, Waldek S, Packman S, Ludington E, Viereck C, Kirk J, Yu J, Benjamin ER, Johnson F, Lockhart DJ, Skuban N, Castelli J, Barth J, Barlow C, Schiffmann R: Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N Engl J Med, 375: 545-555, 2016
44. Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Stork S, Voelker W, Ertl G, Wanner C, Strotmann J: Long-term effects of enzyme replacement therapy on fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation, 119: 524-529, 2009
45. Pieroni M, Camporeale A, Della Bona R, Sabini A, Cosmi D, Magnolfi A, Bolognese L: Progression of Fabry cardiomyopathy despite enzyme replacement therapy. Circulation, 128: 1687-1688, 2013
46. Lombardo M, Alli C, Broccolino M, Ferrari S, Montemurro L, Zaini G, Zanni D: Long-term effects of angiotensin-converting enzyme inhibitors and calcium antagonists on the right and left ventricles in essential hypertension. Am Heart J, 134: 557-564, 1997
47. Tahir H, Jackson LL, Warnock DG: Antiproteinuric therapy and fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol, 18: 2609-2617, 2007
48. Kramer J, Bijnens B, Stork S, Ritter CO, Liu D, Ertl G, Wanner C, Weidemann F: Left Ventricular Geometry and Blood Pressure as Predictors of Adverse Progression of Fabry Cardiomyopathy. PLoS One, 10: e0140627, 2015
49. Eng CM, Germain DP, Banikazemi M, Warnock DG, Wanner C, Hopkin RJ, Bultas J, Lee P, Sims K, Brodie SE, Pastores GM, Strotmann JM, Wilcox WR: Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med, 8: 539-548, 2006
50. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, Gregoratos G, Klein G, Moss AJ, Myerburg RJ, Priori SG, Quinones MA, Roden DM, Silka MJ, Tracy C, Smith SC, Jr., Jacobs AK, Adams CD, Antman EM, Anderson JL, Hunt SA, Halperin JL, Nishimura R, Ornato JP, Page RL, Riegel B, Blanc JJ, Budaj A, Dean V, Deckers JW, Despres C, Dickstein K, Lekakis J, McGregor K, Metra M, Morais J, Osterspey A, Tamargo JL, Zamorano JL, American College of Cardiology/American Heart Association Task F, European Society of Cardiology Committee for Practice G, European Heart Rhythm A, Heart Rhythm S: ACC/AHA/ESC 2006 Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation, 114: e385-484, 2006
51. Verocai F, Clarke JT, Iwanochko RM: Case report: Long-term outcome post-heart transplantation in a woman with Fabry’s disease. J Inherit Metab Dis, 33 Suppl 3: S385-387, 2010
52. Ersozlu S, Desnick RJ, Huynh-Do U, Canaan-Kuhl S, Barbey F, Genitsch V, Muller T, Cheetham M, Flammer A, Schaub S, Nowak A: Long-Term Outcomes of Kidney Transplantation in Fabry Disease. Transplantation, 201852.
53. Eng CM et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis 2007;30(2):184-92


info@herz+gefäss
- Vol. 9
- Ausgabe 1
- Januar 2019