Die pulmonale Hypertonie (PH) ist eine pathophysiologische Entität, welche bei vielen Krankheiten auftritt und zu schwerwiegenden pulmonalen und kardiovaskulären Symptomen führen kann. Durch gute Achtsamkeit kann eine PH rechtzeitig vermutet, die entsprechenden Abklärungen durchgeführt und eine, für die verschiedenen Ätiologien gezielte Therapie eingeleitet werden. Diesem Umstand tragen die neuen Richtlinien der Europäischen Gesellschaft für Kardiologie und der europäischen Respiratory Society Rechnung, indem sie einen Hauptschwerpunkt auf die Erkennung und den Algorithmus zur Diagnose legen (1). Weitere wichtige Neuerungen hat es in den hämodynamischen Definitionen, der Klassifikation und der Risikostratifizierung der Patienten mit PH gegeben. Eine Aktualisierung der gegenwärtigen verfügbaren Therapien und auch deren empfohlener Einsatz sind in den Richtlinien dargestellt. Insgesamt bringen die neuen Richtlinien viele Neuheiten. Auf die wichtigsten davon will dieser Artikel hinweisen.
Pulmonary hypertension (PH) is a pathophysiological entity that occurs in many diseases and can lead to serious pulmonary and cardiovascular symptoms. With good awareness, PH can be suspected and in due time the necessary workup performed and the appropriate therapy initiated for the various etiologies. The new guidelines of the European Society of Cardiology and the European Respiratory Society take this into account by placing a major emphasis on detection and the algorithm for diagnosis (1). There have been other important innovations in hemodynamic definitions, classification, and risk stratification of patients with PH. An update of currently available therapies and also their recommended use are presented in the guidelines. Overall, the new guidelines bring many novelties. This article aims to point out the most important of them.
Key Words: pulmonary hypertension, ESC guidelines
Neue hämodynamische Definition der pulmonalen Hypertonie
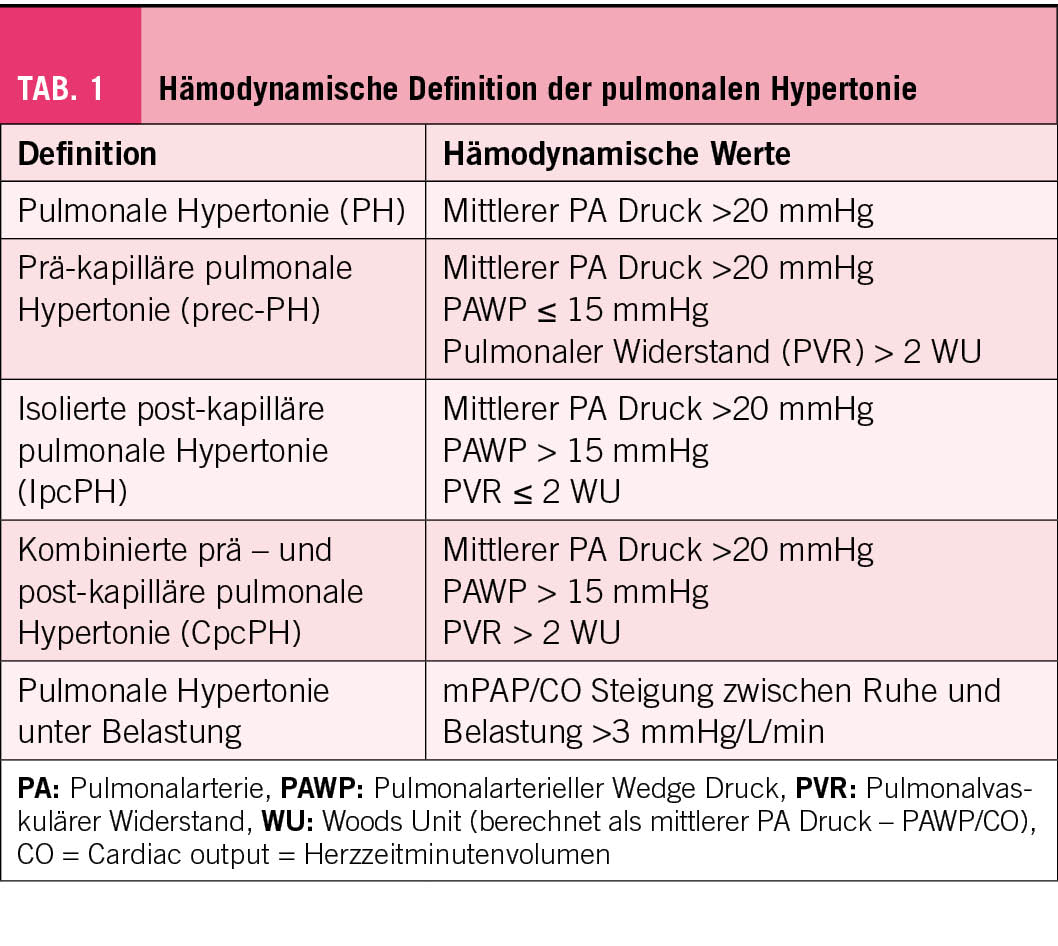
Eine der wichtigsten Neuerungen ist die neue hämodynamische Definition der PH. Die Schwelle von 25 mmHg für den mittleren pulmonalen Druck (mPAP) wurde auf 20 mmHg gesenkt (Tab. 1). Diese Definition wurde am sechsten Weltsymposium für PH erarbeitet und jetzt von den Fachgesellschaften in ihre Richtlinien übernommen (2). Diese tiefere Grenze des (mPAP) von <20 mmHg wird damit begründet, dass ein normaler pulmonaler Druck noch tiefer liegt und dass eine Erhöhung des mittleren PA-Druckes >20 mmHg zu einer schlechten Prognose im Langzeitverlauf bei der idiopathischen und der chronisch thrombo-embolischen pulmonalen Hypertonie führt. Die prä-kapilläre und die post-kapilläre PH werden unterschieden aufgrund des mittleren pulmonalarteriellen Wedge Druckes (PAWP) und des pulmonalvaskulären Widerstands (PVR) gemessen mittels Wood Units (PVR in WU= mittlerer pulmonaler Druck – PAWP geteilt durch das Herzzeitminutenvolumen). Beträgt der PAWP ≤15 mmHg und der PVR >2 Woods Units handelt es sich um eine präkapilläre PH. Beträgt der PAWP >15 mmHg und der PVR ≤2 Wood Units, so liegt eine post-kapilläre PH vor. Gegenüber 2015 wird also der PVR wieder zur Klassifikation der PH verwendet. Hingegen ist die noch in den 2015 verwendete Identifizierung der reinen präkapillären PH mittels des diastolischen Druckgradienten (diastolischer pulmonal-arterieller Druck – mittlerer PAWP) von 7 mmHg fallen gelassen worden. Die kombinierte prä- und post-kapilläre pulmonale Hypertonie wird neu mit einem PVR von >2 WU definiert (Tab. 1). Wichtig ist zu bemerken, dass die neue Schwelle für die Definition einer PH die Empfehlungen für den Therapiebeginn nicht beeinflusst haben. Es gibt nämlich keine Evidenz für die Wirksamkeit einer spezifischen Therapie bei mPAP-Werten <25 mmHg.
Neu sind erstmals auch diagnostische Kriterien für die belastungsabhängige PH definiert worden. Es muss der mittlere PA-Druck (mPAP) und das Herzzeitminutenvolumen (HZV) in Ruhe und unter Belastung gemessen werden. Wenn der Anstieg des Quotienten mPAP/HZV von Ruhe zu Belastung >3 mmHg beträgt, spricht man von einer belastungsabhängigen pulmonalen Hypertonie. Praktisch bedeutet dies, dass bei vermuteter belastungsabhängiger PH ein Rechtsherzkatheter in Ruhe und unter Belastung durchgeführt werden muss. Das ist ein grosser Aufwand, aber dürfte insbesondere bei PatientInnen mit Herzinsuffizienz mit erhaltener Auswurffraktion hilfreich sein bei der Evaluation der Ursachen der Dyspnoe.
Klassifikation der pulmonalen Hypertonie
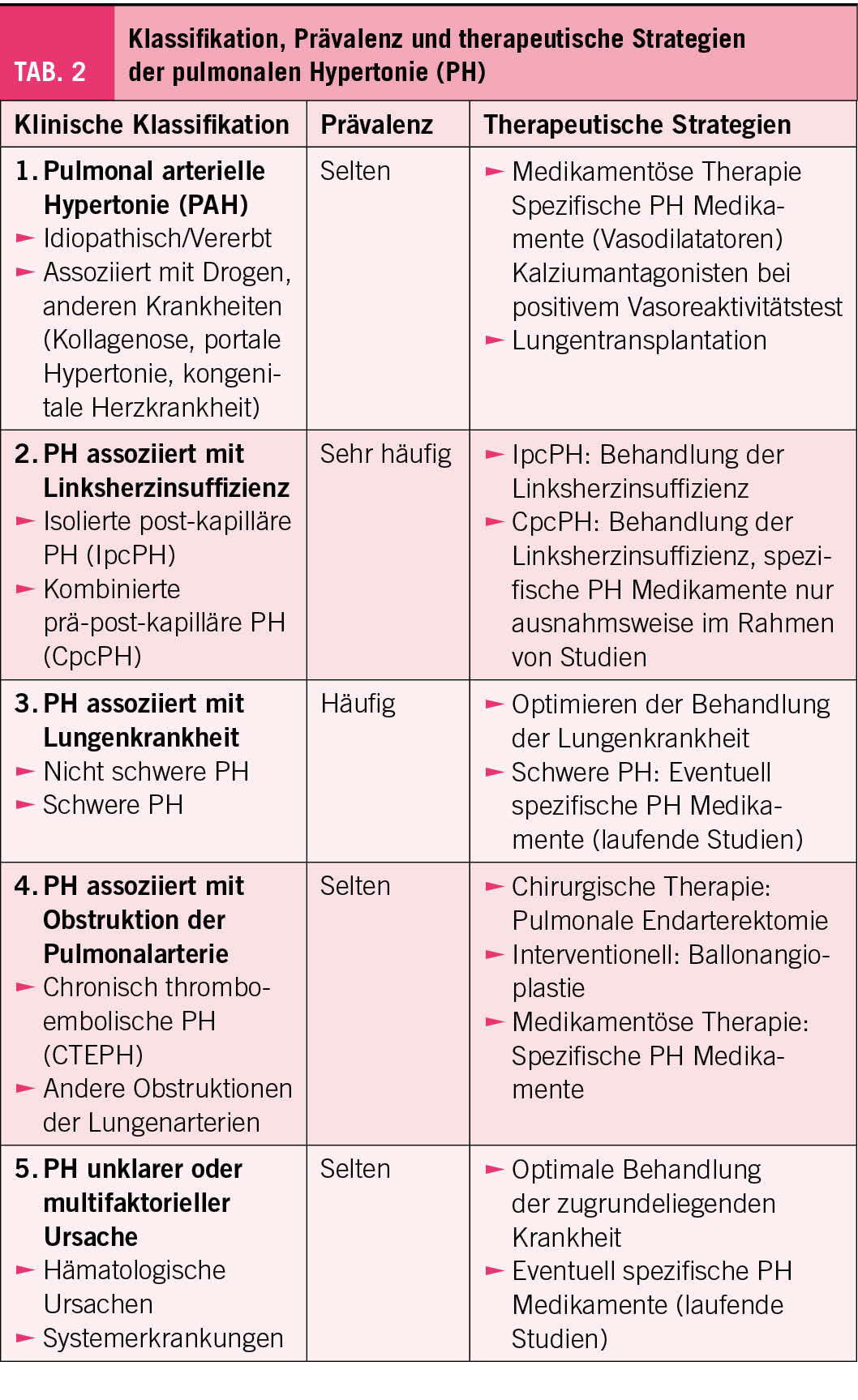
Die Grundstruktur der klinischen Klassifikation der PH in fünf Gruppen wurde beibehalten (Tab. 2). Mit Abstand am häufigsten wird die PH durch eine Linksherzinsuffizienz verursacht, gefolgt von der PH assoziiert mit Lungenkrankheiten. Alle anderen Ätiologien sind selten. Bei der Gruppe 1, dh. der pulmonal-arteriellen Hypertonie wird bei der Untergruppe idiopathische pulmonal-arterielle Hypertonie neu unterschieden zwischen «Responders» und der «Non-responders» aufgrund der Testung der Vasoreaktivität. «Responders» können initial mit Kalziumanatagonisten behandelt werden und haben eine etwas bessere Prognose. Des Weiteren wurde die veno-okklusive Ätiologie neu der pulmonal-arteriellen Hypertonie (Gruppe 1) als Untergruppe zugeteilt.
Diagnostische Abklärung
a. Verdachtsdiagnose und allgemeines Vorgehen
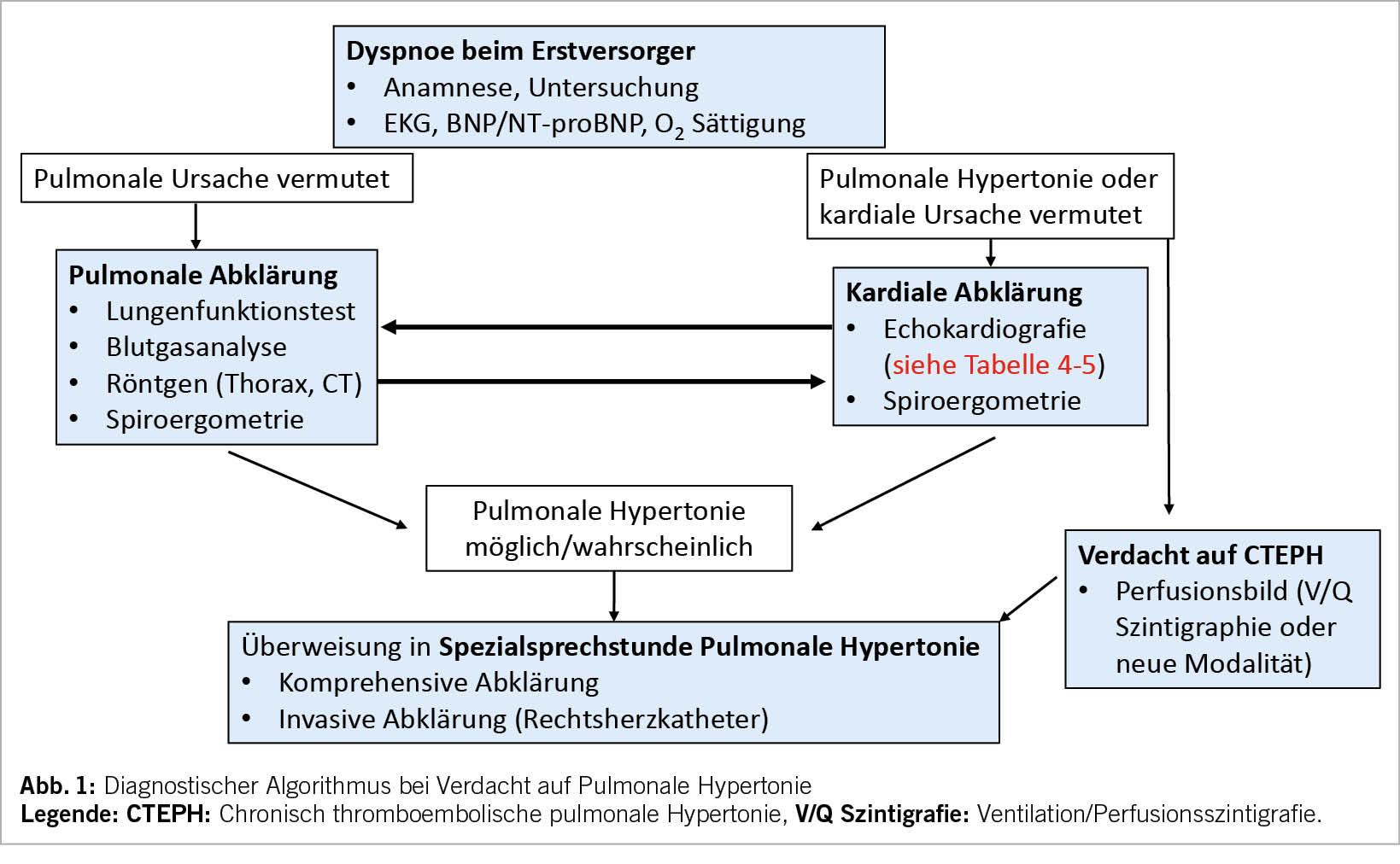
Die Empfehlungen für die Abklärung bei Verdacht auf eine PH sind vollkommen neu strukturiert worden und folgen dem in der klinischen Praxis gängigen Patientenpfad. Die Empfehlungen stellen die Echokardiografie ganz in den Vordergrund der Abklärung. Bei Patienten mit Dyspnoe sollte der erstuntersuchende Arzt an eine PH als seltene Ursache der Dyspnoe denken (Abb. 1). Bei Verdacht auf eine PH soll, wie bei Verdacht auf eine kardial bedingte Dyspnoe, der Patient dem Kardiologen für eine Echokardiografie zugewiesen werden. Ergibt die Echokardiografie die mögliche oder wahrscheinliche Diagnose einer PH soll eine umfassende Abklärung, die alle Spezialuntersuchungen, welche zur Evaluation der Ätiologie nötig sind, erfolgen. In den allermeisten Fällen gehört zur Sicherung der Diagnose auch eine invasive Messung der Hämodynamik im Rechtsherzkatheter. Ebenfalls soll bei Patienten, bei den eine pulmonale Krankheit als Ursache der Dyspnoe vermutet wurde und bei denen anlässlich der pulmonalen Abklärung eine PH vermutet wird, einer Echokardiografie durchgeführt werden. Umgekehrt sollen Patienten, bei denen aufgrund der Echokardiografie die Wahrscheinlichkeit für eine PH tief ist, den Pneumologen zur weiteren Abklärung überwiesen werden (Abb. 1, Tab. 4). Die chronische thromboembolische pulmonale Hypertonie (CTEPH) soll frühzeitig gesucht werden, mit einem Perfusionsbild, z.B. V/Q Szintigraphie, SPECT-CT oder CT-Thorax mit i.v. Kontrastmittel und Dual-Energy Protokoll. Risikofaktoren für eine CTEPH sind Lungenembolie oder Thrombose in der Vorgeschichte, Tumorerkrankung, Splenektomie und hämatologische Erkrankungen.
b. Abklärung mittels Echokardiografie
Es gibt aufgrund der multiplen Ätiologien für eine PH keinen einzelnen echokardiografischen Parameter, aufgrund dessen die Diagnose einer PH zweifelsfrei gestellt werden kann. Vielmehr erlaubt die Echokardiografie mittels Messen der Flussgeschwindigkeit des trikuspidalen Regurgitationsjets (TVR) und einer sorgfältigen Suche der indirekten Zeichen einer PH die Wahrscheinlichkeit der Diagnose PH anzugeben (Tab. 4 und 5). Geschwindigkeiten <2,8 m/s sprechen gegen und Geschwindigkeiten >3,4 m/s für das Vorliegen einer PH (Tab. 4). Aus der TVR lässt sich der systolische pulmonale Druck abschätzen (TVR2x4). Dazu müsste aber der rechts-atriale Füllungsdruck bekannt sein, respektive abgeschätzt werden. Da die Abschätzung des Füllungsdrucks sehr variable Werte ergibt, empfehlen die Guidelines, dass nicht der geschätzte systolische Pulmonaldruck, sondern alleine die Geschwindigkeit des TVR zur Beurteilung der Wahrscheinlichkeit einer PH verwendet wird.
Zusätzlich zur Flussgeschwindkigkeit des TVR Jets geben die indirekten Zeichen für eine PH Hinweise für die Wahrscheinlichkeit einer PH. Neu wird die Bewegung des Trikuspidalanuöus als Mass für die Kontraktion des rechten Ventrikels, als indirektes Zeichen in die Empfehlungen aufgenommen. Dabei wird die tricuspidal anulus plane systolic excursion (TAPSE) gemessen und dem systolischen Pulmonaldruck gegenübergestellt. Wenn das Verhältnis TAPSE/sPAP <0.55mm/mm beträgt, spricht das für das Vorliegen einer PH (Tab. 5).
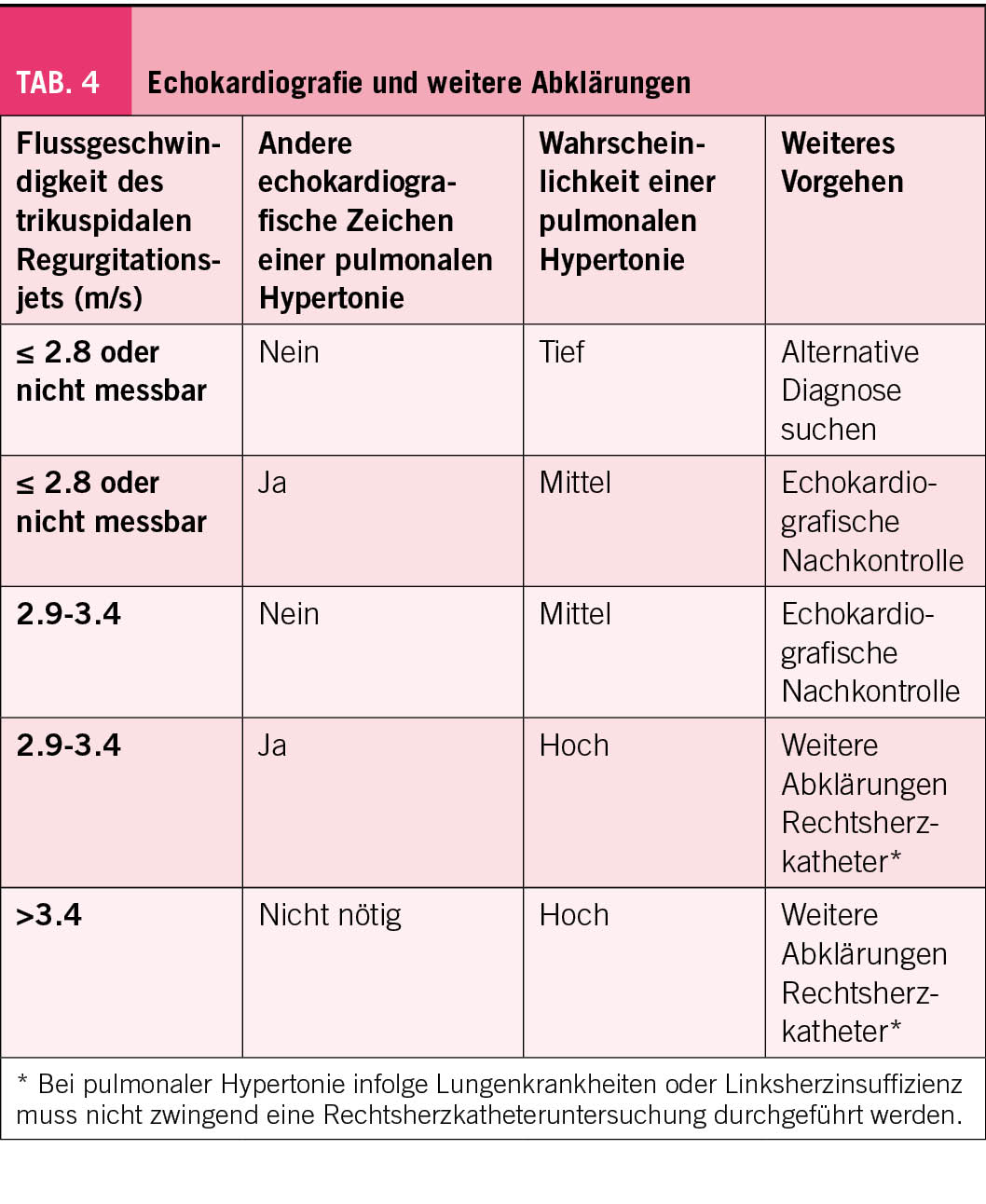
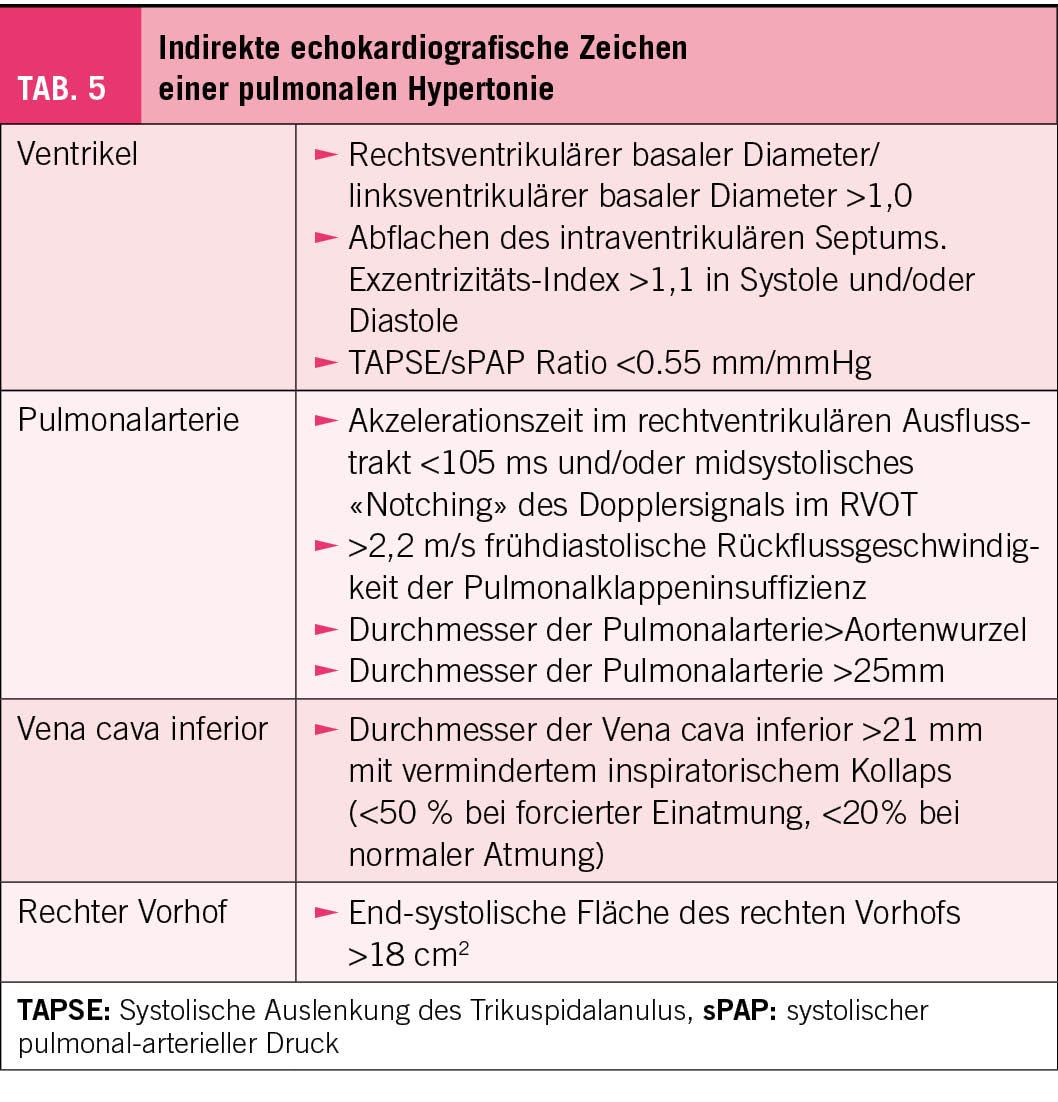
Zu beachten ist, dass, im Gegensatz zur hämodynamischen Neudefinition der PH aufgrund der invasiven Messung (mPAP >20 mmHg), der Wert von >2.8 m/s der TVR als Schwelle für die Verdachtsdiagnose für eine PH nicht verändert wurde. Es hat sich nämlich gezeigt, dass eine TVR >2.8 m/s bei 25%-35% der Allgemeinpopulation und bei >45% der aus klinischen Indikationen durchgeführten Echokardiografien vorliegt (3). Die leicht erhöhten pulmonal-arteriellen Drücke werden durch erhöhte links-atriale Füllungsdrück, Steifigkeit der Pulmonalarterien, und Remodeling der Pulmonalgefässe, wie sie insbedondere im Alter, bei Frauen und bei metabolischen Krankheiten vorkommen, verursacht (3). Der systolische Pulmonaldruck steigt mit dem Alter an und Werte bis 36 mmHg (dh. eine TVR bis 3 m/s) sind bei Personen >60 Jahre normal (4).
c. Bestätigung mittels Rechtsherzkatheter
Die Rechtsherzkatheteruntersuchung bleibt der Goldstandard für die Diagnose und Klassifikation der PH (Tab. 1). Wenn mittels Echokardiografie die Verdachtsdiganose einer PH gestellt ist, sollte eine Rechtsherzkatheruntersuchung an einem Zentrum durchgeführt werden. Die invasive Untersuchung muss eine sorgfältige Messung der Hämodynamik und der Sättigungen im pulmonalen und systemischen Kreislauf beinhalten. Eine Prüfung der Vasoreaktivität ist nur bei Patienten mit pulmonal-arterieller Hypertonie (Gruppe 1) angezeigt, um diejenigen Patienten zu finden, welche mittels Kalziumantagonisten behandelt werden können. Bei den anderen Ätiologien ist eine Vasoreaktivität nicht zu erwarten und hätte keine therapeutischen Konsequenzen. Bei Patienten mit Dyspnoe und Verdacht auf eine PH aber normaler Hämodynamik in Ruhe empfehlen die Guidelines neu eine Rechtsherzuntersuchung unter Belastung durchzuführen. Dies wird am ehesten bei Patienten mit einer Herzinsuffizienz mit erhaltener Auswurffraktion hilfreich sein. Eine Rechtsherzuntersuchung unter Belastung kann auch prognostische und funktionelle Informationen bei Patienten mit Verdacht auf eine pulmonal-arterielle Hypertonie (Gruppe 1) oder bei der CTEPH liefern.
Risikostratifikation
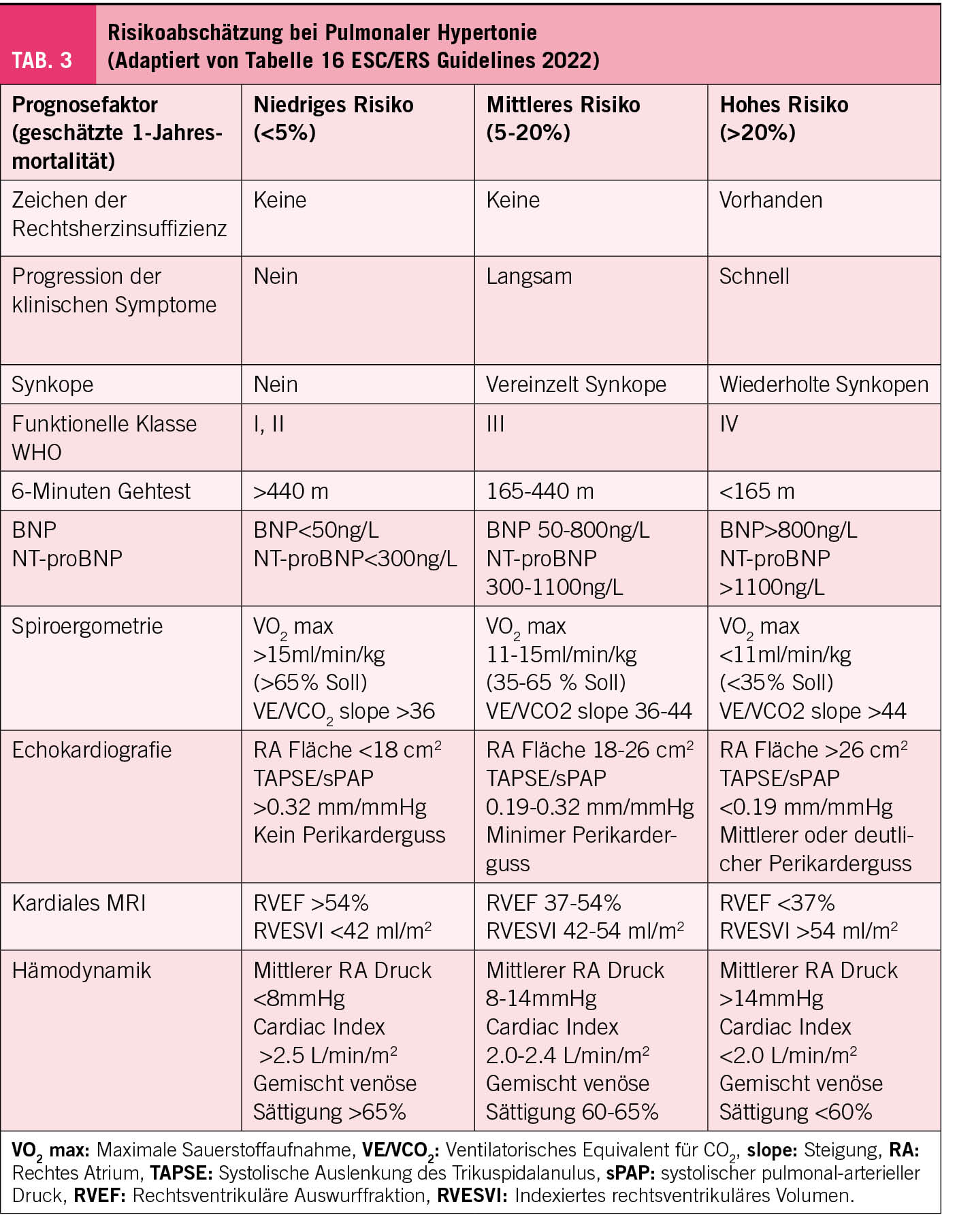
Einhergehend mit der Diagnosestellung in der Spezialsprechstunde für PH muss die Ätiologie durch verschiedene Spezialuntersuchungen eruiert werden. Im Anschluss daran soll eine Risikostratifizierung erfolgen (Tab. 3). Die Risikostratifizierung ist in den neuen Guidelines verfeinert und ausgedehnt worden. Diese Risikostratifizierung ist gut validiert für Patienten mit pulmonal-arterieller Hypertonie (Gruppe 1). Für die anderen Ätiologie besteht keine vergleichbare zuverlässige Risikoabschätzung. Es wird zwischen einem tiefen (Mortalitätsrisiko innerhalb eines Jahres <5%), einem mittleren (Mortalitätsrisiko 5-20%) und einem hohen Risiko (Mortalität >20% innert einem Jahr) unterschieden. Neben den klinischen Symptomen und Zeichen wird das Risiko mittels der funktionellen Tests (6-Minuten-Gehtest, Spiroergometrie), dem natriuretischen Peptid, der invasiv gemessenen Hämodynamik und neu auch mittels der Befunde im MRI festgelegt. Von den echokardiografischen Parameter (5) werden neu auch die Werte der systolischen Auslenkung des Trikuspidalanulus (TAPSE) einbezogen. Das Risiko an der PH zu versterben, bestimmt die Intensität der initialen Behandlung.
Behandlung
Die Behandlung der PH richtet sich nach deren Ätiologie (Tab. 2). Für die Patienten mit pulmonal-arterieller Hypertonie der Gruppe 1 (idiopathische PH, vererbte PH, PH assoziiert mit Medikamenten, Drogen, anderen Krankheiten) stehen die spezifischen vasodilatierenden Medikamente, welche über die Endothelin, die NO oder die Prostazyklin vermittelte Vasodilatation wirken, zur Verfügung. Neu wird die Therapie aufgrund des Vorliegens von anderen kardiopulmonalen Komorbiditäten modifiziert. Wenn andere kardiopulmonale Erkrankungen vorliegen, wird primär eine Monotherapie eingesetzt, wenn nicht, wird empfohlen die PH-Medikamente von Anfang an in einer Zweierkombination zu verabreichen. Beim Vorliegen eines hohen Risikos soll rasch eine Dreiertherapie begonnen werden (mit zusätzlich i.v. oder subkutan Prostazyklin Analogen). Nach 3-6 Monaten soll die Therapie überprüft und angepasst werden aufgrund einer neuen Risikoevaluation, welche die Veränderung der Dyspnoe, des 6-Minuten-Gehtests und des natriuretischen Peptids beinhaltet.
Eine orale Antikoagulation wird nicht mehr generell empfohlen. Die diuretische Therapie spielt eine wichtige Rolle in der Rechtsherzinsuffizienz, welche mit einer Hypervolämie, reduzierter renaler Durchblutung und der Aktivierung des Renin-Angiotensin-Aldosteron Systems assoziiert ist. Dabei können die Schleifendiuretika gut mit anderen Diuretika, insbesondere Spironolactone kombiniert werden. Die chronische Hypoxämie verschlechtert die PH durch die zusätzliche hypoxische Vasokonstriktion. Deshalb wird eine Dauersauerstoffstherapie bereits bei einem PaO2 von <8 kPa empfohlen.
Die Empfehlungen bei Patienten mit CTEPH folgen den Entwicklungen der interventionellen und medikamentösen Therapien. Bei diesen Patienten ist eine lebenslange Antikoagulation nötig. Eine Beurteilung der Operabilität und Empfehlung für die Therapie soll im Rahmen einer interdisziplinären Besprechung stattfinden, in der Schweiz wurde vor mehreren Jahren ein nationales CTEPH Board eingeführt, welches online monatlich die Fälle diskutiert.
Wenn der Patient operabel ist, dann soll er im Hinblick auf eine pulmonale Endarteriektomie evaluiert werden. Eine solche ist möglich, wenn proximale fibrotische Obstruktionen vorliegen. Bei distalen fibrotischen Obstruktionen soll eine Ballonangioplastie der befallenen Pulmonalarterien evaluiert werden. Die Operation wird in Zürich (USZ) durchgeführt, die Ballonangioplastie in Genf (HUG), Bern (Inselspital) und Zürich (USZ).
Für Veränderungen im mikrovaskulären Bereich stehen die spezifischen vasodilatierenden Medikamente zur Verfügung. Die beste Evidenz hat Riociguat, das via Stimulation der löslichen Guanylatzyklase die NO vermittelte Vasodilatation fördert. Schwächere Evidenz hat der Endothelinantagonist Macitentan und wird deshalb erst konditionell als Erstmedikament empfohlen. Die medikamentöse Therapie wird multimodal eingesetzt mit der chirurgischen oder interventionellen Behandlung der CTEPH.
Bei allen anderen Formen der PH wird die Behandlung der Grundkrankheit ohne Einsatz der vasodilatierenden Medikamente empfohlen. Einzig bei der schweren Form der PH aufgrund einer Lungenkrankheit empfehlen die Guidelines den vorsichtigen Einsatz spezifischer Medikamente, am besten im Rahmen von klinischen Studien. Es ist allerding anzumerken, dass in der klinischen Praxis sich zunehmend ältere Patienten (>70 Jahre) mit PH präsentieren. Bei ihnen ist oft eine genaue Unterscheidung in eine ätiologische Klasse nicht möglich, wie zum Beispiel bei Patienten mit Herzinsuffizienz mit gemischt prä- und postkapillärer Hypertonie. Evidenz für einen Nutzen der PH-Medikamente bei solchen Patienten fehlen und wir müssen entsprechende Studien abwarten. Dies gilt insbesondere für die grösste Patientengruppe mit PH assoziiert mit Linksherzinsuffizienz. Bei Patienten mit Herzinsuffizienz mit erhaltener Auswurffraktion hat sich Sildenafil jedenfalls als nicht hilfreich erwiesen (5). Bei Patienten mit Herzinsuffizienz bleibt die Guideline empfohlene Herzinsuffizientherapie die optimale Behandlung.
Copyright bei Aerzteverlag medinfo AG
Stadtspital Zürich Waid
Pneumologie
Leiterin Sprechstunde für pulmonale Hypertonie
Tièchestrasse 99
8037 Zürich
Stadtspital Zürich Triemli
Klinik für Kardiologie
Birmensdorferstrasse 497
8063 Zürich
franz.eberli@triemli.zuerich.ch
Die Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618-731.
2. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. European Respiratory Journal. 2019;53(1).
3. Jankowich M, Maron BA, Choudhary G. Mildly elevated pulmonary artery systolic pressure on echocardiography: bridging the gap in current guidelines. Lancet Respir Med. 2021;9(10):1185-91.
4. Ferrara F, Rudski LG, Vriz O, Gargani L, Afilalo J, D’Andrea A, et al. Physiologic correlates of tricuspid annular plane systolic excursion in 1168 healthy subjects. Int J Cardiol. 2016;223:736-43.
5. Hoendermis ES, Liu LC, Hummel YM, van der Meer P, de Boer RA, Berger RM, et al. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J. 2015;36(38):2565-73.


info@herz+gefäss
- Vol. 13
- Ausgabe 2
- April 2023