Anlässlich der Jahresversammlung 2019 der Schweizerischen Gesellschaft für Kardiologie fand ein Symposium zum Thema «Pulmonale Hypertonie» statt. Dieser Bericht fasst zwei wichtige Beiträge zu diesem Thema zusammen: das Einstiegsreferat von Prof. Dr. med. Markus Schwerzmann sowie die von Prof. Dr. med. Otto Schoch präsentierten Therapieoptionen bei Pulmonaler Hypertonie.
Einstiegsreferat zum Krankheitsbild
Eine Übersicht zur Definition der pulmonalen Hypertonie, zu den verschiedenen Klassen der pulmonalen Hypertonie, zu den Todesursachen von Patienten mit pulmonaler Hypertonie und zu den prognostischen Markern gab

Prof. Dr. med. Markus Schwerzmann, Bern. Der Blutdruck in den beiden Kreisläufen, Körperkreislauf und Lungenkreislauf, ist unterschiedlich. Während er im grösseren Körperkreislauf, in welchem das Blut einen grösseren Widerstand überwinden muss, etwa 120mmHg auf 80mmHg (Mittelwert ca 93mmHg) beträgt, sind die mittleren Werte im Lungenkreislauf nur etwa 20 auf 8 mmHg (Mittelwert 15 mmHg).
Der transpulmonale Druckgradient (TPG), definiert durch die Differenz zwischen dem mittleren pulmonal arteriellen Druck (PAP) und dem linken Vorhofdruck (allgemein geschätzt durch den pulmonalen Kapillarkegeldruck), wird für den Nachweis einer intrinsischen pulmonalen Gefässerkrankung bei Linksherz-Zuständen im Zusammenhang mit einem erhöhten pulmonalen Venendruck empfohlen
Der pulmonal-kapilläre Verschlussdruck (PCWP, pulmonary capillary wedge pressure) wird gemessen, indem ein Herzkatheter in eine periphere Vene (z.B. Jugular- oder Oberschenkelvene) eingeführt und dann in den rechten Vorhof, rechten Ventrikel, Lungenarterie und in einen Ast der Lungenarterie vorgeschoben wird. Normal ist ein TPG von 7mmHg, eine pulmonale Hypertonie liegt ab einem Wert von 15mmHg vor.
Ätiologie der pulmonalen Hypertonie
Mögliche Mechanismen sind
- Erhöhter Fluss: Zustände mit hohem Output, kongenitale Herzerkrankung
- Erhöhter Widerstand: Lungenerkrankung, Verdünnung der Lungengefässe, idiopathische pulmonal arterielle Hypertonie, Lungenembolie und ihre Konsequenzen. Der Referent verglich die Mechanismen mit einem Gartenschlauch, der voll aufgedreht oder gestaut werden kann.
- Erhöhter PCWP, rheumatische Herzkrankheit, Linksherz-Krankheit (Aortenstenose, Herzinsuffizienz, Mitralinsuffizienz, etc.).
Diagnose – Echokardiographie / TTE
Es kann nur der systolische Blutdruck gemessen werden, nicht der mittlere Blutdruck. Transpulmonale Gradienten können nicht berechnet werden.
Üblicherweise ist der mittlere PAP = 0.6 x systolischer PAP. Systolischer PAP > 40 mmHg bedeutet pulmonale Hypertonie.
Ursachen für pulmonale Hypertonie weltweit
In Europa sind es zu je 48% Linksherz-Krankheit und Lungenkrankheit und zu 4% andere Ursachen wie idiopathische, Bindegewebserkrankung, HIV, portale Hypertonie, kongenitale Herzerkrankung. Im mittleren Osten sind es Linksherz-Krankheit zu 50%, Lungenkrankheit zu 40%, in Afrika zu 40% Linksherzkrankheit und zu 30% Lungenkrankheit.
Klinische Klassifikation der pulmonalen Hypertonie
Die pulmonale Hypertonie (PH) wird in 5 verschiedene Gruppen eingeteilt:
Gruppe 1: pulmonale arterielle Hypertonie (PAHT)
Idiopathisch, genetisch, mit pulmonal arterieller Hyper-
tonie assoziierte Krankheit, KHK, HIV, Sklerodermie,
Leberkrankheit
Gruppe 2: PH wegen Linksherz Krankheit
Gruppe 3: PH wegen Lungenkrankheit
Gruppe 4: PH wegen chronischer Lungenembolie
Gruppe 5: PH infolge unklarer Mechanismen
z.B. Sarkoidose, Splenektomie, Sichelzellanämie
Diagnostische Schritte bei Verdacht auf pulmonale Hypertonie
Bei Verdacht auf eine pulmonale Hypertonie gilt es zunächst die pulmonale Hypertonie zu bestätigen (TPG, PCWG) und anschliessend nach der Ätiologie zu suchen: Herzkrankheit? Lungenkrankheit? Lungenembolie? Rheumatische Erkrankung? HIV? Lebererkrankung?
Prognose mit pulmonaler Hypertonie – die ausfallende rechte Herzkrammer
Bei pulmonaler Hypertonie passt sich die rechte Herzkammer der zunehmenden Gefässbelastung an, indem sie die Kontraktilität erhöht, um den Fluss aufrecht zu erhalten. Die ventrikuloarterielle Kopplung bedeutet, dass sich das Hubvolumen wenig ändert und gleichzeitig der ventrikuläre Wirkungsgrad erhalten bleibt. Letztendlich entsteht eine Phase, in der eine ventrikuläre Dilatation stattfindet, um die Reduzierung des Hubvolumens zu begrenzen, mit Entkopplung und damit verbundener erhöhter Wandbeanspruchung. Da die Kopplung durch Hypertrophie bis zum Endstadium der Erkrankung aufrechterhalten wird, wenn die fortschreitende Dilatation beginnt, ist das rechtsventrikuläre Volumen der wesentliche Parameter zur Messung bei der Nachsorge von Patienten mit pulmonaler Hypertonie.
Fazit
- Die pulmonale Hypertonie ist häufig auf eine Linksherz-Erkrankung oder pulmonale Lungenerkrankung zurückzuführen
- Die Diagnose erfordert eine Katheterisierung
o mittlerer pulomal-arterieller Druck
o PCWP - Verschiedene Krankheiten führen zu peripherer arterieller pulmonaler Hypertonie/pulmonaler Hypertonie
- Die Prognose hängt von der ventrikulären Funktion ab
- Prognosefaktoren sind funktionelle Kapazität, ventrikuläre Funktion, Herzzeitvolumen
Therapieoptionen bei pulmonaler Hypertonie

Eine umfassende Diagnostik ist Voraussetzung für eine gezielte Therapie, stellte Pof. Dr. med. Otto Schoch, St. Gallen eingangs fest. Er erwähnte die verschiedenen Klassen der pulmonalen Therapie.
Die Therapieoptionen bei PH Klasse 2
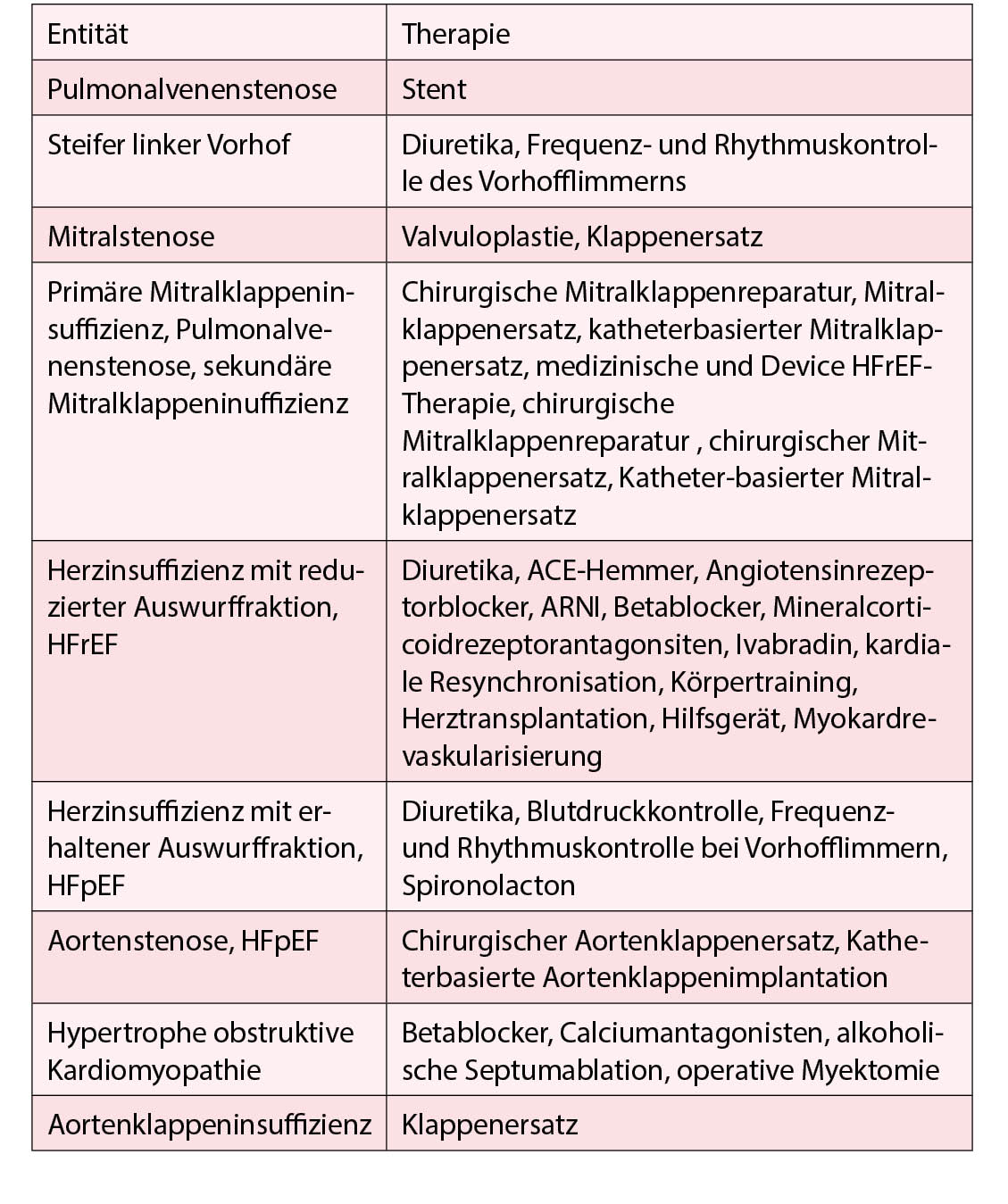
Pulmonale Hypertonie im Zusammenhang mit der linksseitigen Herzkrankheit (PH-LHD) ist die häufigste Form der pulmonalen Hypertonie. Bei Patienten mit linksseitiger Herzerkrankung ist das Vorhandensein von pulmonaler Hypertonie typischerweise ein Marker für fortgeschrittenere Erkrankungen, schwerwiegendere Symptome und schlechtere Prognosen. Die Therapieoptionen der verschiedenen Entitäten der PH Klasse 2 sind in der Tabelle enthalten.
Die SERAPHIN-Studie mit dem Endothelinrezeptorantagonisten Macicentan zeigte, dass die Langzeitbehandlung der PAH mit Macicentan die Morbidität und Mortalität verringern kann.
Therapieoptionen bei PH Klasse 3
Diese beinhaltet Pulmonale Hypertonie infolge Lungenkrankheiten und/oder Hypoxie. Sie umfasst folgende Krankheitsbilder: Chronisch obstruktive Lungenkrankheit (COPD), interstitielle Lungenkrankheit, andere Lungenkrankheiten mit gemischtem restriktivem und obstruktivem Muster, Schlafatmungsstörungen, Alveoläre Hypoventilationsstörungen, chronische Höhenexposition und Entwicklungsstörungen der Lungen.
Die Therapieoptionen bei PH Klasse 3 je nach Krankheitsbild umfassen
- bei COPD:
- Raucherentwöhnung, Rehablilitation, Inhalation (Beta-Agonisten, Anticholinergika), Steroide, Antibiotika (gegen Exazerbationen), Langzeit-Oxygen, Reduktion des Lungenvolumens, Lungentrasplantation;
- bei Interstitiellen Lungenkrankheiten:
- Langzeit Oxygen, immunsuppressive und antifibrotische Wirkstoffe, Lungentransplantation;
- bei anderen Lungenkrankheiten (chronische Lungenfibrose mit Emphysem): Raucherentwöhnung, Langzeit-Oxygen, Lungentransplantation
- Bei Schlafatmungsstörungen:
CPAP, Lebensstiländerungen (Gewichtsreduktion etc.), Unterkiefervorschubgeräte; - bei alveolären Hypoventilationsstörungen:
nichtinvasive Ventilation, Gewichtsreduktion (Hypoventilation bei Übergewicht); - bei chronischer Höhenexposition:
Verschiebung auf geringere Meereshöhe, Langzeit-Oxygen; - bei Entwicklungsabnomalitäten: Chirurgische Behandlung erwägen.
Therapieoptionen bei PH Klasse 4
Diese umfasst die Chronisch Embolische Pulmonale Hypertonie (CTEPH) und es steht die Operation (Pulmonale Endarteriektomie) im Vordergrund. Falls technisch nicht operabel, sollte eine Zweitmeinung eingeholt werden und eine gezielte medikamentöse Therapie in Erwägung gezogen werden. Bei dieser PH-Klasse wurde Riociguat (Adempas®) als erste Substanz zugelassen. Adempas® ist auch zur Behandlung der pulmonal-arteriellen Hypertonie zugelassen.
Therapieoptionen bei PH Klasse 1, pulmonal arterielle Hypertenie (PAH)
Die PAH kann idiopathisch, vererbt (BMPR2-Muttion, andere Mutationen), Medikament- oder Toxin-induziert oder mit verschiedenen Krankheiten, wie Bindegewebserkrankung, HIV Infektion, Portalhypertonie, kongenitaler Herzkrankheit oder Schistosomiasis assoziiert sein. «Wenn es eine Revolution in der medizinischen Therapie in den letzten Jahrzehnten gibt, ist es die Behandlung der PAH», so Sergio Harari (Eur Respir Rev 2016;35:361-363). Der Referent nennt verschiedene medikamentöse Therapien und die Empfehlungen gemäss der WHO FC (Galié N. et al Eur Heart J 2015;doi10.1093:1-58). Macicentan zu Sildenafil, Riociguat zu Bosentan, oder Selexipag zu einem ERA und/PDE5i werden darin als Klasse-I/B-Empfehlungen erwähnt.
Fazit
- Therapieoptionen bei pulmonaler Hypertonie sind abhängig von der Diagnosekategorie: vollständiges Assessment inklusive Herzkatheter, Lungenfunktion, Radiologie, VQ-Scan, Ergospirometrie, Schlafstudien etc.
- PH Klasse 2 (kardial) und 3 (pulmonal) kausale Therapie, keine Medikamente für pulmonal-arterielle Hypertonie
- PH Klasse 4 (CTEPH) primär Operation evaluieren
- PAH Klasse 1: Spezifische Therapie aus 3 Substanzklassen, initial oder bei unbefriedigendem Ansprechen im Verlauf Kombination
- Basistherapie: O2- Supplementation und Rehabilitation nicht vergessen
Quelle: SGK Jahrestagung Interlaken, 19.-21. Juni 2019
riesen@medinfo-verlag.ch
