Meningeome sind die häufigsten primären intrakraniellen Tumoren im Erwachsenenalter, die aus den Zellen der Arachnoidea entstehen. Obwohl viele Meningeome zufällig entdeckt werden und asymptomatisch bleiben, erfordern einige aufgrund neurologischer Symptome eine Behandlung. Risikofaktoren umfassen hormonelle Einflüsse, ionisierende Strahlen und genetische Prädispositionen wie Neurofibromatose Typ 2. Die Diagnose erfolgt primär durch bildgebende Verfahren, während die Behandlung meist chirurgisch ist, ergänzt durch Strahlentherapie bei unvollständiger Resektion oder aggressivem Verhalten. Neue molekulare Klassifikationen und Therapien sind im Fokus der aktuellen Forschung, um die Prognose und Behandlungseffizienz zu verbessern.
Meningiomas are the most common primary intracranial tumors in adults, originating from the cells of the arachnoid. While many meningiomas are incidentally discovered and remain asymptomatic, some require treatment due to neurological symptoms. Risk factors include hormonal influences, ionizing radiation, and genetic predispositions such as Neurofibromatosis Type 2. Diagnosis is primarily through imaging techniques, with treatment typically being surgical, supplemented by radiotherapy in cases of incomplete resection or aggressive behavior. Current research focuses on new molecular classifications and therapies to improve prognosis and treatment efficacy.
Key words: Meningioma, Arachnoid Cells, Neurofibromatosis Type 2, Radiotherapy, Molecular Classification
Einleitung und Epidemiologie von Meningeomen
Meningeome gehen aus Zellen der Arachnoidea hervor, einer spinnwebartigen Schicht der Hirnhäute zwischen Dura mater und Pia mater. Im Erwachsenenalter sind Meningeome die mit Abstand häufigsten primären intrakraniellen Tumoren (1). In einer populationsbasierten Magnetresonanztomographiestudie wurden gar Prävalenzen für Meningeome um 1.6 % in der Allgemeinbevölkerung berichtet und in Autopsiestudien von bis zu 3 % (2, 3). Allerdings bedürfen nur wenige zufällig diagnostizierte Meningeome einer Behandlung. Die jährliche Inzidenz histopathologisch diagnostizierter Meningeome, d.h. solcher, die zumeist aufgrund einer neurologischen Symptomatik diagnostiziert und reseziert wurden, liegt um 8.5/100 000 und steigt mit zunehmendem Alter bis hin zu einer Inzidenz von deutlich über 40/100 000 unter den über 75-jährigen (1).
Hormonelle Einflüsse und Risikofaktoren
Prämenopausale Frauen sind etwa doppelt so häufig von symptomatischen Meningeomen betroffen wie Männer (1). Diese Beobachtung sowie Assoziationen mit Schwangerschaft, Brustkrebs, Adipositas oder hormoneller Kontrazeption spiegeln eine Rolle von Progesteron bei der Entstehung eines Teils der Meningeome wider (4–7). Meningeome können Rezeptoren für Östrogen als auch für Progesteron exprimieren, wobei Progesteronrezeptoren deutlich häufiger und in etwa 70–80 % der Tumoren nachgewiesen werden können (8, 9). Passend zu einer Wachstumsstimulation durch hohe Progesteronspiegel gibt es Fallberichte von Patientinnen, bei denen es während einer Schwangerschaft zu einem massiven Tumorwachstum und postpartal zu einem spontanen Regress kam (9, 10). So erklärt sich auch, dass Meningeome, die bei älteren Patientinnen als Zufallsbefunde diagnostiziert werden, oftmals kein Wachstum mehr in der Verlaufsbeobachtung zeigen. Ferner wird dieser Zusammenhang durch Berichte über die Assoziation von Fertilitätsbehandlungen und dem Auftreten von Meningeomen (11–13) sowie das Auftreten von Meningeomen bei Mann-zu-Frau Transgendern unter Hormontherapie (14) unterstützt. Dieses Wissen sollte dazu führen, dass Hormonbehandlungen wie Hormonersatztherapie oder hormonale orale Kontrazeptiva bei Meningeompatienten und -patientinnen sehr kritisch geprüft werden.
Genetische Prädisposition und Neurofibromatose Typ 2
Bei Betroffenen der Neurofibromatose Typ 2 treten bis zum 70. Lebensjahr in > 80 % der Fälle Meningeome auf, bei einem mittleren Alter bei Diagnosestellung um das 30. Lebensjahr, Fehlen von Geschlechterunterschieden und oft multifokalem Wachstum (15). In einer populationsbasierten Langzeituntersuchung in Manchester (Vereinigtes Königreich) bedurfte während 30 Jahren jedoch weniger als jeder dritte Patient/jede dritte Patientin mit bildgebendem Verdacht auf ein Meningeom auch einer Resektion wegen neurologischer Symptome (16).
Ionisierende Strahlung als Risikofaktor
Zu den etablierten Risikofaktoren für das Auftreten von Meningeomen zählt zudem die Exposition gegenüber ionisierenden Strahlen, z. B. im Rahmen einer Strahlentherapie der Neuraxis zur Behandlung von Leukämien oder Tumorerkrankungen im Kindesalter. Populationsbasierte Studien in Australien und im Vereinigten Königreich zeigten, dass bereits das Dosisäquivalent einer einzigen, nach aktuellen Standards durchgeführten nativen Computertomographie des Kopfes im Kindes- und Jugendalter, mit einer vielfachen Erhöhung des Risikos für die Entstehung von Meningeomen und anderen Tumoren des Zentralnervensystems einhergeht (17, 18). Meningeome können mit einer Latenz von bis zu mehreren Jahrzehnten nach Strahlenexposition auftreten, oftmals multifokal und mit ungünstigerer Prognose als sporadisch auftretende Meningeome (19).
Klinische Präsentation und Bildgebung
Klinisch machen sich Meningeome zumeist mit Kopfschmerzen oder durch neurologische Symptome wie Sehstörungen, neurokognitive Einschränkungen, sensomotorische Defizite oder Epilepsie bemerkbar. Die Verdachtsdiagnose wird im Rahmen einer Computertomographie oder einer Magnetresonanztomographie gestellt (Abbildung 1). Typische radiologische Zeichen umfassen die extraaxiale, duraständige Lokalisation mit einem «dural tail», eine homogene Kontrastmittelaufnahme, einen hyperintensen Saum zwischen Kontrastmittel und Hirnparenchym sowie gelegentliche Kalzifikation und angrenzende Hyperostose. Das Vorliegen eines Ödems deutet im Gegensatz zu einer Kalzifikation eher auf ein aktives Tumorwachstum hin. Neben dem typischen «spiegeleiartigen» Erscheinungsbild kommen auch sogenannte en-plaque und seltener intraaxial infiltrierend wachsende Meningeome vor, die nicht immer sicher von leptomeningeal disseminierten oder intraaxialen Tumoren wie Hirnmetastasen oder Gliomen abzugrenzen sind. Auch wenn die bildgebenden Zeichen des Meningeoms oftmals eindeutig erscheinen, kann die definitive Diagnosestellung daher nur gewebebasiert erfolgen.
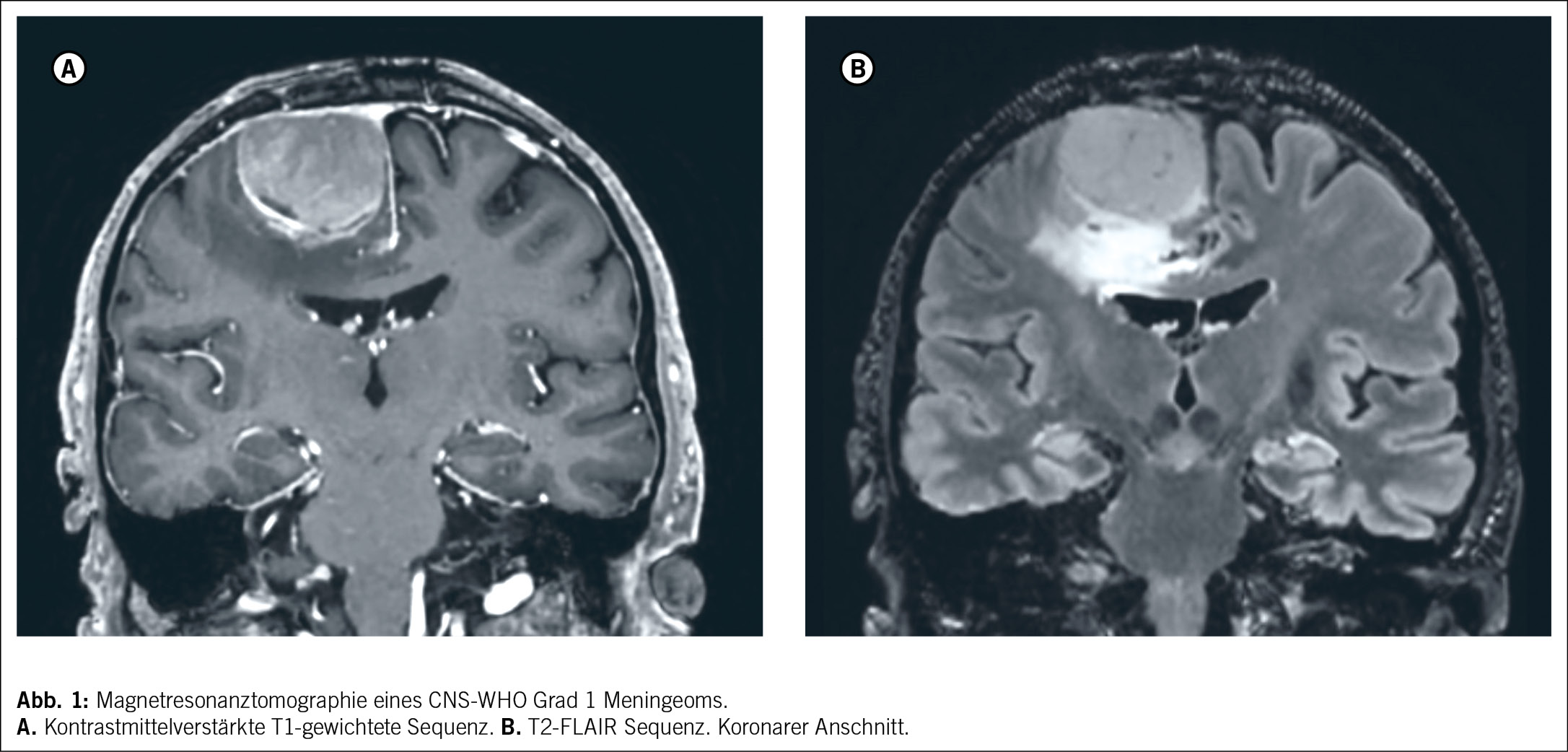
Chirurgische Behandlung und Resektionstechniken
Die chirurgische Resektion erfolgt in aller Regel bei symptomatischen Patient/-innen oder bei deutlicher Grössenprogression im Rahmen einer «watch-and-wait»-Strategie. Klassischerweise werden Meningeome über eine Schädelöffnung (Kraniotomie) entfernt. Wenn möglich, werden dabei auch die Hirnhäute, an denen der Tumor ansetzt, mit entfernt. Der Duradefekt wird dann mit autologem Gewebe (z.B. Galea oder Faszia lata) oder mit einem künstlichen Duraersatz gedeckt. Das Resektionsausmass wird üblicherweise mit der Simpson-Klassifikation angegeben, die ein Spektrum von vollständiger Tumorentfernung mit zusätzlicher Entfernung des duralen Ansatzes und gegebenenfalls der beteiligten Knochen (Grad I) bis zu einer erweiterten Biopsie (Grad V) reicht (20). Mit Hilfe dieser Klassifikation soll unter anderem das Risiko für ein Rezidiv eingeschätzt werden. Allerdings stammt die Simpson-Klassifikation aus dem Jahr 1957 und ist damit deutlich vor der Mikrochirurgie und modernen bildgebenden Verfahren wie der Computertomographie oder der Magnetresonanztomographie entstanden. Da Meningeome meist sehr stark vaskularisiert sind, ist ein wichtiger Teil der chirurgischen Strategie die frühe Unterbindung der Blutzufuhr mittels Koagulation der duralen Gefässe, die den Tumor versorgen. Dadurch soll der intraoperative Blutverlust gemindert werden. Bei Meningeomen, deren zuführende Gefässe erst am Ende des Eingriffs zugänglich werden (z. B. Meningeome an der Schädelbasis), kann auch eine OP-vorbereitende Embolisation über einen intraarteriellen Katheter in Erwägung gezogen werden.
Strahlentherapie und ergänzende Behandlungen
Die Prognose ist im Anschluss an eine radiographische Komplettresektion insgesamt günstig. Dauerhafte neurologische Symptome und Beeinträchtigungen der Lebensqualität sind jedoch auch bei solchen Patient/-innen häufig, bei denen nach einer chirurgischen Resektion kein Tumorwachstum mehr beobachtet wird (21). Zudem bedarf etwa jeder vierte Meningeompatient /jede vierte Meningeompatientin einer zusätzlichen Behandlung jenseits der initialen Resektion (22). Die Behandlungsoptionen sind dabei beschränkt auf erneute chirurgische Resektionen von Rezidiven oder einer Strahlentherapie, z. B. nach inkompletter Resektion oder bei Vorliegen von prognostisch ungünstigen histo- und molekularpathologischen Zeichen (Abbildung 2) (23). Die Strahlentherapie erfolgt zumeist als intensitätsmodulierte Strahlentherapie mit 50−55 Gy in Einzeldosen von 1.8–2 Gy. Nicht resektable, kleine Meningeome können auch als stereotaktische Strahlenchirurgie mit einer Einzeldosis von 14–16 Gy effektiv behandelt werden. Bei enger Nachbarschaft zu kritischen anatomischen Strukturen wie dem Nervus opticus kommen darüber hinaus auch Protonen-basierte Protokolle zum Einsatz (23).
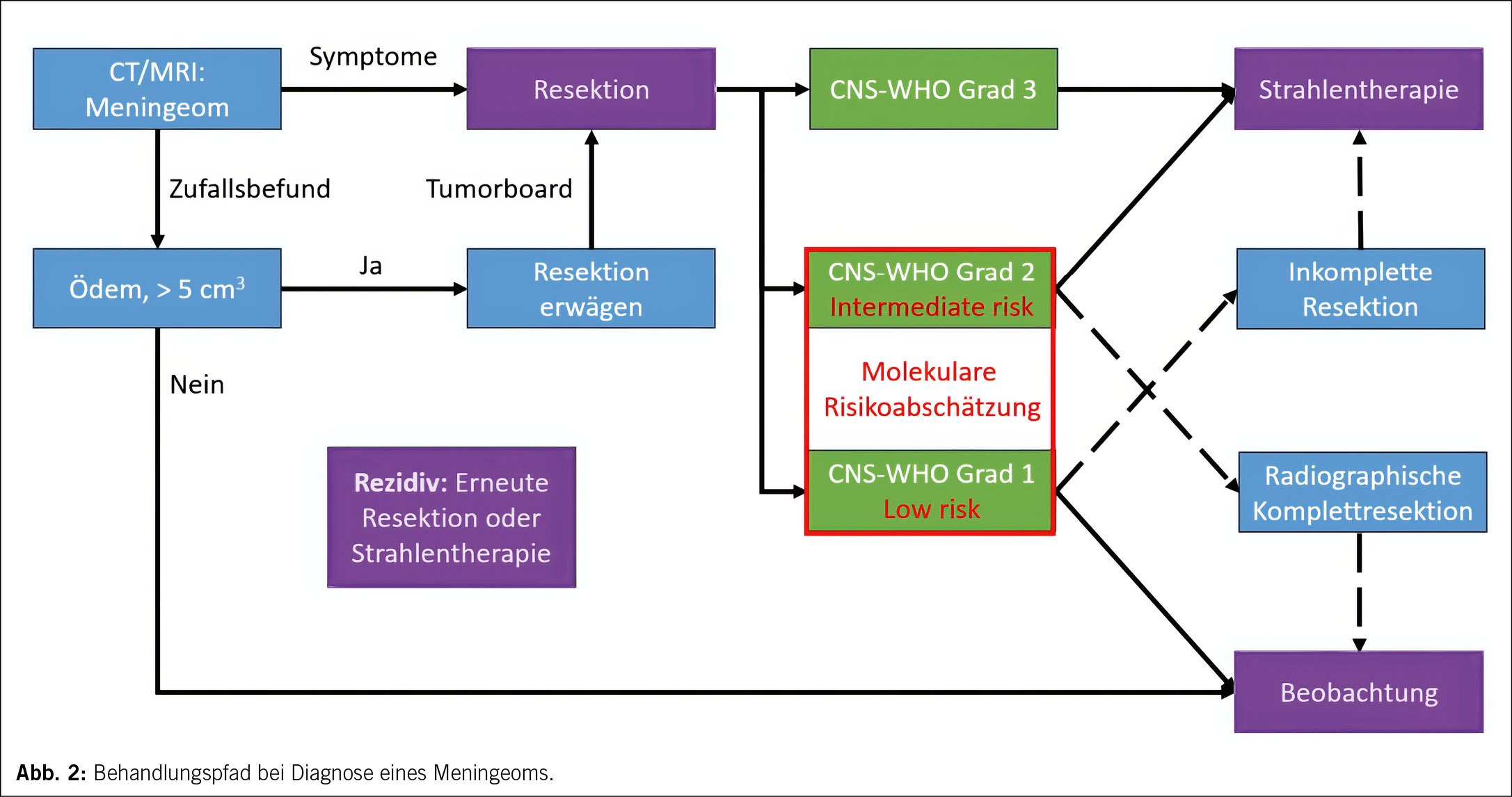
Risikoklassifikation und Prognose
Für die Frage, ob postoperativ eine Strahlentherapie erfolgen sollte oder nicht, ist eine zuverlässige prognostische Klassifikation unabdingbar. Die Einteilung von Meningeomen erfolgt nach den Kriterien der 2021 revidierten Klassifikation von Tumoren des Zentralnervensystems der Weltgesundheitsorganisation in drei Risikoklassen, die CNS-WHO Grade 1–3 (24). Diese Klassifikation erfolgt primär histopathologisch, wobei etwa 80 % der Meningeome den sechs Subtypen des benignen CNS-WHO Grad 1, knapp 20 % den drei Subtypen des intermediären CNS-WHO Grad 2 und einige wenige den drei Subtypen des seltenen, malignen CNS-WHO Grad 3 zugeordnet werden. Intraaxiales, invasives Wachstum und eine erhöhte Proliferationsrate sind auch in histopathologisch benignen Subtypen prognostisch ungünstige Kriterien, die eine Zuordnung zum CNS-WHO Grad 2 bedingen. Zudem wurden 2021 homozygote Deletionen des CDKN2A/B Genlokus sowie Mutationen im TERT-Genpromotor als prognostisch sehr ungünstige molekulare Alterationen in die CNS-WHO Klassifikation eingeführt, die unabhängig vom histopathologischen Erscheinungsbild definierend für den CNS-WHO Grad 3 sind (24).
Die prognostische Zuordnung gelingt insbesondere an der Grenze zwischen CNS-WHO Grad 1 und Grad 2 nicht immer. Das heisst, ein Teil der als benigne klassifizierten Meningeome rezidiviert trotz einer radiographischen Komplettresektion, während ein Teil der Meningeome, die dem prognostisch ungünstigeren Grad 2 zugeordnet wurden, nach vollständiger Resektion auch ohne anschliessende Strahlentherapie kein erneutes Tumorwachstum zeigen. Um die Indikationsstellung für eine Strahlentherapie zu verbessern, wurden daher zunehmend auch objektivierbare, molekulare Klassifikationskonzepte jenseits der Histopathologie entwickelt. So wurden prognostisch günstige Subtypen identifiziert, die durch Mutationen in den Genen AKT1, SMO, KLF4 oder TRAF7 charakterisiert sind (25). Die überwiegende Mehrzahl der Meningeome weist jedoch Mutationen im NF2-Gen auf. Diese prädestinieren für die zusätzliche Akkumulation chromosomaler Verluste, von denen insbesondere 1p, 6q, 10q, 14q und 22q mit ungünstiger Prognose assoziiert sind (26). Die DNA-Methylierungsanalyse resezierter Meningeomgewebe bietet eine breit verfügbare, günstige Möglichkeit, sowohl chromosomale Aberrationen zu identifizieren als auch eine Einteilung in prognostische Methylierungssubgruppen vorzunehmen (27). Kürzlich wurde basierend auf der klinisch-molekularen Analyse von mehreren Kohorten mit insgesamt über 3000 Patient/-innen ein prognostischer «Score» vorgeschlagen, der Histopathologie, Gensequenzierung, chromosomalen Aberrationen und Methylierungsklasse integriert und so eine deutlich präzisere Voraussage der Prognose erlaubt, als die CNS-WHO Klassifikation (26).
Laufende Studien und therapeutische Entwicklungen
Die Frage, welchen Patient/-innen eine Strahlentherapie empfohlen werden sollte, wird auch durch die internationale, randomisierte Phase-2-Studie «ROAM» adressiert, die aktive Verlaufskontrollen mit einer unmittelbar postoperativ angeschlossenen Strahlentherapie nach vollständiger Tumorresektion eines CNS-WHO Grad 2 Meningeoms vergleicht. Die Rekrutierung der Studie ist abgeschlossen, erste Ergebnisse werden Ende 2026 erwartet (28). Post hoc Analysen dieser Studie werden mit Blick auf eine nach aktuellen Standards durchgeführte Strahlentherapie zudem Hinweise auf deren Wirksamkeit innerhalb molekularer Subgruppen sowie auf mögliche Effekte auf Neurokognition und Lebensqualität liefern.
Das Fehlen wirksamer, etablierter Systemtherapien stellt für die Behandlung von Patient /-innen mit Meningeom, bei denen die Strahlentherapie nicht zum gewünschten Erfolg führt, eine besondere Herausforderung dar. Bislang konnte die Wirksamkeit unterschiedlichster Systemtherapien nicht in kontrollierten klinischen Studien nachgewiesen werden (29). Insbesondere zeigten verschiedene zytotoxische Ansätze bestenfalls geringe Wirksamkeit. Eine extensive Immunsuppression des Tumormikromilieus und das Vorhandensein nur weniger Protein verändernder Mutationen machen Meningeome zudem zu ungünstigen Kandidaten für immuntherapeutische Ansätze (30). Eine Behandlung mit dem antiangiogenen Tyrosinkinasehemmer Sunitinib zeigte in einer unkontrollierten Phase II Studie in 36 Patient/-innen mit CNS-WHO Grad 2 oder Grad 3 Meningeom zeitlich begrenzte Tumorstabilisierung in einem Teil der behandelten Patient/-innen (31). Auch die Kombination antiangiogenen monoklonalen Antikörpers Bevacizumab und mTor-Hemmers Everolimus wurde in einer unkontrollierten Phase-II-Studie untersucht und konnte die Erkrankung in 12 Patient/-innen mit CNS-WHO Grad 2 oder Grad 3 Meningeom im Median für 22 Monate stabilisieren (32). Eine molekular zielgerichtete Behandlung mit 177Lutethium-gebundenen Agonisten des Meningeom-assoziierten Rezeptors SSTR2 zeigte in retrospektiven Analysen ebenfalls nur geringe Effektivität in einem Teil der Patient/-innen mit Meningeom (33). All diese Ansätze bedürfen jedoch einer Bestätigung in prospektiven, randomisierten Studien.
Zusammengefasst stellt die Entwicklung wirksamer, nebenwirkungsarmer Therapien jenseits von Chirurgie und Strahlentherapie eine Schlüsselherausforderung für das Management von Meningeompatient /-innen mit ungünstigen Verläufen dar. Nach vollständiger Resektion eines Meningeoms werden jedoch in der weit überwiegenden Mehrzahl der Patient /-innen günstige Verläufe ohne Rezidive beobachtet.
Copyright Aerzteverlag medinfo AG
Klinik für Neurochirurgie, Kantonsspital St. Gallen,
Rorschacher Strasse 95
9007 St. Gallen
Klinik für Neurologie,
Kantonsspital Winterthur
Brauerstrasse 15
8400 Winterthur
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Price, M. et al. CBTRUS Statistical Report: American Brain Tumor Association & NCI Neuro-Oncology Branch Adolescent and Young Adult Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016-2020. Neuro Oncol 26, iii1-iii53 (2024). https://doi.org:10.1093/neuonc/noae047
2. Johnson, M. D. & Abu-Farsakh, S. Clinicopathologic features of incidental meningiomas: A review of the literature and the University of Rochester autopsy experience. Clin Neuropathol 38, 118-121 (2019). https://doi.org:10.5414/NP301160
3. Vernooij, M. W. et al. Incidental findings on brain MRI in the general population. N Engl J Med 357, 1821-1828 (2007). https://doi.org:10.1056/NEJMoa070972
4. Degeneffe, A., De Maertelaer, V., De Witte, O. & Lefranc, F. The Association Between Meningioma and Breast Cancer: A Systematic Review and Meta-analysis. JAMA Netw Open 6, e2318620 (2023). https://doi.org:10.1001/jamanetworkopen.2023.18620
5. Muskens, I. S. et al. Body mass index, comorbidities, and hormonal factors in relation to meningioma in an ethnically diverse population: the Multiethnic Cohort. Neuro Oncol 21, 498-507 (2019). https://doi.org:10.1093/neuonc/noz005
6. Niedermaier, T. et al. Body mass index, physical activity, and risk of adult meningioma and glioma: A meta-analysis. Neurology 85, 1342-1350 (2015). https://doi.org:10.1212/WNL.0000000000002020
7. Pettersson-Segerlind, J. et al. The risk of developing a meningioma during and after pregnancy. Sci Rep 11, 9153 (2021). https://doi.org:10.1038/s41598-021-88742-2
8. Commins, D. L., Atkinson, R. D. & Burnett, M. E. Review of meningioma histopathology. Neurosurg Focus 23, E3 (2007). https://doi.org:10.3171/FOC-07/10/E3
9. Lusis, E. A. et al. Meningiomas in pregnancy: a clinicopathologic study of 17 cases. Neurosurgery 71, 951-961 (2012). https://doi.org:10.1227/NEU.0b013e31826adf65
10. Chakravarthy, V. et al. Houdini Tumor: Case Report and Literature Review of Pregnancy-Associated Meningioma. World Neurosurg 114, e1261-e1265 (2018). https://doi.org:10.1016/j.wneu.2018.03.187
11. Frassanito, P., De Bonis, P., Mattogno, P. P., Novello, M. & Anile, C. Hormonal therapy for fertility and huge meningioma: a purely random association? Acta Neurol Belg 112, 299-301 (2012). https://doi.org:10.1007/s13760-012-0046-9
12. Motegi, H. et al. Hemorrhagic onset of rhabdoid meningioma after initiating treatment for infertility. Brain Tumor Pathol 29, 240-244 (2012). https://doi.org:10.1007/s10014-012-0088-y
13. Patterson, A. & Elashaal, A. Fast-Growing Meningioma in a Woman Undergoing Fertility Treatments. Case Rep Neurol Med 2016, 3287381 (2016). https://doi.org:10.1155/2016/3287381
14. Ter Wengel, P. V., Martin, E., Gooren, L., Den Heijer, M. & Peerdeman, S. M. Meningiomas in three male-to-female transgender subjects using oestrogens/progestogens and review of the literature. Andrologia 48, 1130-1137 (2016). https://doi.org:10.1111/and.12550
15. Goutagny, S. et al. Long-term follow-up of 287 meningiomas in neurofibromatosis type 2 patients: clinical, radiological, and molecular features. Neuro Oncol 14, 1090-1096 (2012). https://doi.org:10.1093/neuonc/nos129
16. Forde, C. et al. Disease course of neurofibromatosis type 2: a 30-year follow-up study of 353 patients seen at a single institution. Neuro Oncol 23, 1113-1124 (2021). https://doi.org:10.1093/neuonc/noaa284
17. Mathews, J. D. et al. Cancer risk in 680,000 people exposed to computed tomography scans in childhood or adolescence: data linkage study of 11 million Australians. BMJ 346, f2360 (2013). https://doi.org:10.1136/bmj.f2360
18. Pearce, M. S. et al. Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: a retrospective cohort study. Lancet 380, 499-505 (2012). https://doi.org:10.1016/S0140-6736(12)60815-0
19. Umansky, F., Shoshan, Y., Rosenthal, G., Fraifeld, S. & Spektor, S. Radiation-induced meningioma. Neurosurg Focus 24, E7 (2008). https://doi.org:10.3171/FOC/2008/24/5/E7
20. Simpson, D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry 20, 22-39 (1957). https://doi.org:10.1136/jnnp.20.1.22
21. Wirsching, H. G., Morel, C., Roth, P. & Weller, M. Socioeconomic burden and quality of life in meningioma patients. Qual Life Res 29, 1801-1808 (2020). https://doi.org:10.1007/s11136-020-02461-1
22. Mastall, M. et al. Survival of brain tumour patients with epilepsy. Brain 144, 3322-3327 (2021). https://doi.org:10.1093/brain/awab188
23. Goldbrunner, R. et al. EANO guideline on the diagnosis and management of meningiomas. Neuro Oncol 23, 1821-1834 (2021). https://doi.org:10.1093/neuonc/noab150
24. Louis, D. N. et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23, 1231-1251 (2021). https://doi.org:10.1093/neuonc/noab106
25. Preusser, M., Brastianos, P. K. & Mawrin, C. Advances in meningioma genetics: novel therapeutic opportunities. Nat Rev Neurol 14, 106-115 (2018). https://doi.org:10.1038/nrneurol.2017.168
26. Maas, S. L. N. et al. Integrated Molecular-Morphologic Meningioma Classification: A Multicenter Retrospective Analysis, Retrospectively and Prospectively Validated. J Clin Oncol 39, 3839-3852 (2021). https://doi.org:10.1200/JCO.21.00784
27. Sahm, F. et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol 18, 682-694 (2017). https://doi.org:10.1016/S1470-2045(17)30155-9
28. Jenkinson, M. D. et al. The ROAM/EORTC-1308 trial: Radiation versus Observation following surgical resection of Atypical Meningioma: study protocol for a randomised controlled trial. Trials 16, 519 (2015). https://doi.org:10.1186/s13063-015-1040-3
29. Mair, M. J., Berghoff, A. S., Brastianos, P. K. & Preusser, M. Emerging systemic treatment options in meningioma. J Neurooncol 161, 245-258 (2023). https://doi.org:10.1007/s11060-022-04148-8
30. Wirsching, H. G. & Weller, M. Immunotherapy for Meningiomas. Adv Exp Med Biol 1416, 225-234 (2023). https://doi.org:10.1007/978-3-031-29750-2_17
31. Kaley, T. J. et al. Phase II trial of sunitinib for recurrent and progressive atypical and anaplastic meningioma. Neuro Oncol 17, 116-121 (2015). https://doi.org:10.1093/neuonc/nou148
32. Shih, K. C. et al. A phase II trial of bevacizumab and everolimus as treatment for patients with refractory, progressive intracranial meningioma. J Neurooncol 129, 281-288 (2016). https://doi.org:10.1007/s11060-016-2172-3
33. Seystahl, K. et al. Somatostatin receptor-targeted radionuclide therapy for progressive meningioma: benefit linked to 68Ga-DOTATATE/-TOC uptake. Neuro Oncol 18, 1538-1547 (2016). https://doi.org:10.1093/neuonc/now060


info@onco-suisse
- Vol. 14
- Ausgabe 6
- September 2024