
MDS sind klonale Bluterkrankungen des älteren Menschen und werden vorwiegend bei Personen im Alter über 70 Jahre diagnostiziert. In europäischen Ländern beträgt die alters-standardisierte Inzidenzrate 2-3 pro 100’000 Patientenjahre mit einer zweifach höheren Inzidenz bei Männern als bei Frauen. Die einzige Ausnahme stellt dabei das MDS mit del(5q) dar, welche eine weibliche Prädominanz hat (1-4). Aufgrund der demographischen Alterung und der zunehmenden diagnostischen Möglichkeiten muss man in Zukunft davon ausgehen, dass die Entität der MDS zu einer der häufigsten hämatologischen Neoplasien aufsteigen wird, mit relevanter Auswirkungen auf die Gesundheitsversorgung (1).
MDS sind heterogene Erkrankungen, die durch sequenzielle Ansammlung von genetischen Läsionen in den hämatopoetischen Stammzellen (HSC) verursacht werden (5). Die genetischen Läsionen, die bisher bei MDS identifiziert werden konnten, waren strukturelle Chromosomenaberrationen, die sich durch eine konventionelle Metaphasen-Zytogenetik oder Fluoreszenz-In-Situ Hybridisierung (FISH) nachweisen lassen. Die Analyseverfahren und dadurch auch das Verständnis genetischer Veränderungen in MDS und anderen myeloischen Neoplasien haben sich jedoch in den letzten Jahren rasant weiterentwickelt (6). Dank der Next Generation Sequencing (NGS) ist es nun möglich, rekurrente somatische Driver-Mutationen (RSDM) nachzuweisen. Diese RSDM treten in Genen mit folgenden Funktionen auf: RNA-Splicing, epigenetische Regulation, Transkriptionsfaktoren, Zellzyklus, Kohesinkomplex und Zellsignalling (7 - 11).
Diagnostik und Klassifizierung
Die diagnostischen Empfehlungen bei erwachsenen Patienten mit vermutetem MDS wurden in den ELN-Empfehlungen 2013 zusammengefasst (7). Eine gründliche persönliche Anamnese (symptomatische Anämie, Infekte, Blutungen und insbesondere Expositionen gegenüber Chemo-/Radiotherapie, Pestizide, Insektizide und Lösungsmittel), Familienanamnese (Hinweise für hereditäre Prädisposition bei jüngeren MDS Patienten) sowie eine körperliche Untersuchung (Blutungszeichen, Organomegalien, Lymphadenopathien) stellen eine wichtige Grundlage dar. Zwingende Laboranalysen umfassen die morphologische Beurteilung des peripheren Blutes, des Knochenmarkaspirates/der Biopsie und eine zytogenetische Analyse. Zusätzlich empfohlene Untersuchungen umfassen die FISH (falls Zytogenetik nicht aussagekräftig) sowie die fluoreszenzaktivierte Zellsortierung (FACS). In speziellen Situationen werden zusätzlich auch molekulare Analysen (array-CGH, PCR und NGS) zum Nachweis von kryptischen Gendefekten und RSDMs empfohlen. Für die Klassifizierung der MDS werden die Anzahl von Zell-Linien, die von Zytopenie und Dysplasie betroffen sind, das Vorhandensein von Ringsideroblasten (RS) oder der Mutationen in SF3B1 (das mit RS assoziiert ist), Anzahl Blasten im peripheren Blut oder Knochenmark und das Vorliegen von speziellen MDS-definierenden zytogenetischen Anomalien (zB del 5q) berücksichtigt (Tab. 1) (12).
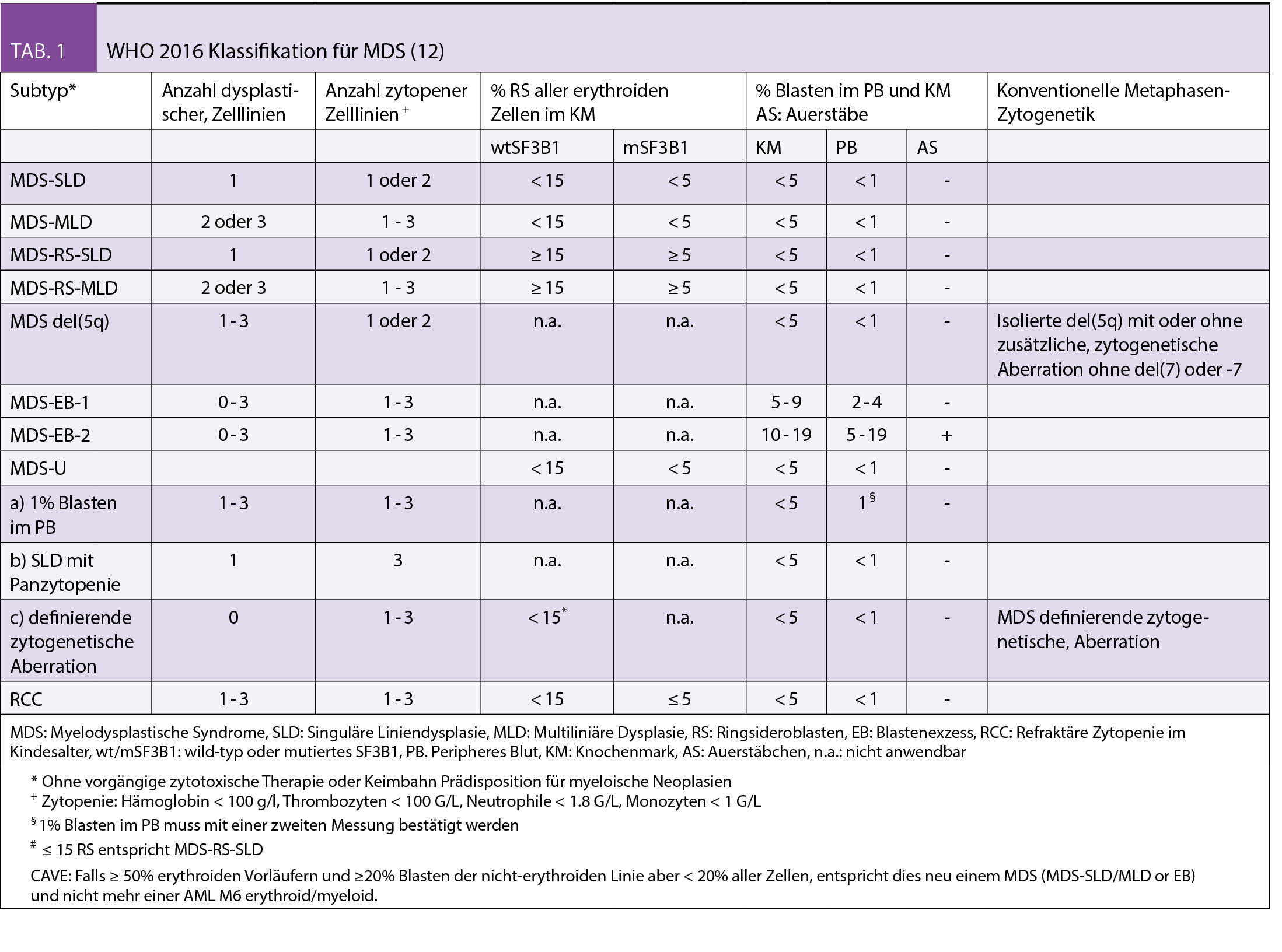
Zytopenien im Alter
Eine ungeklärte Anämie findet sich bei etwa 10 - 15% der Patienten im Alter von > 65 Jahren und es ist nicht immer einfach, reaktive von klonalen Zuständen abzugrenzen. Patienten mit einer Zytopenie, jedoch ohne ausreichende dysplastische Veränderungen oder MDS-definierende zytogenetische Veränderungen, werden als idiopathische Zytopenie unklarer Signifikanz (ICUS) bezeichnet (13). Bei diesen Patienten wird eine Verlaufskontrolle nach 3 - 6 Monaten und ggf. eine Wiederholung der Knochenmarksuntersuchung empfohlen (7). RSDM können mit einer altersabhängigen, erhöhten Häufigkeit bei älteren Patienten (10-20%) nachgewiesen werden. Diese Personen haben normale periphere Blutwerte oder nur eine leichte Zytopenie, welche aber die diagnostischen Kriterien für MDS nicht erfüllen. Diese Zustände werden als klonale Hämatopoiese mit indeterminiertem Potential (CHIP: normale periphere Blutwerte ohne RSDM) (14, 15) oder klonale Zytopenie unklarer Signifikanz (CCUS: Zytopenie mit RSDM) (16) bezeichnet. Die Anwendung der NGS ermöglicht neuerdings Patienten mit reaktiven und klonalen Zuständen zu unterschieden. Auf diese Weise lassen sich Patienten in frühen Stadien einer klonalen Hämatopoiese identifizieren, welche ein erhöhtes Risiko für die Entwicklung einer overten hämatologischen Neoplasie haben.
Allgemeine Überlegungen zum MDS Patientenmanagement
MDS sind sehr heterogene Erkrankungen mit einem sehr variablen natürlichen Verlauf von chronischen, asymptomatischen Zytopenien bis zu einem schnellen Fortschreiten in eine sekundäre AML. Zwei Drittel der Patienten mit MDS sterben an zytopenieassoziierten Komplikationen und ein Drittel erliegt der AML-Progression (17). Das Management basiert auf einer krankheits- und patientenassoziierten Risikostratifizierung. Eine exakte Diagnose und Risikostratifizierung sind für eine korrekte Behandlung daher entscheidend. Patienten mit niedrigem Risiko haben ein medianes Überleben von 3 bis 8 Jahren auf und sterben vorwiegend an zytopenieassoziierte Komplikationen (kardiovaskuläre Ereignisse, Infektionen und Blutungen). Die Ziele einer Behandlung von Patienten mit niedrigem Risiko liegen daher vor allem in der Verbesserung der Lebensqualität, Verringerung von zytopenieassoziierten Komplikationen und einem Hinauszögern eines Progresses in ein höhergradiges MDS (18, 19). Bei MDS Patienten mit hohem Risiko liegt hingegen die mediane Überlebenszeit nur bei 1 bis 3 Jahre und die AML-assoziierte Mortalität steht im Vordergrund. Bei diesen Patienten sollte die Behandlung darauf ausgerichtet sein, die Progression in eine AML hinauszuzögern und das Gesamtüberleben zu verbessern (20, 21).
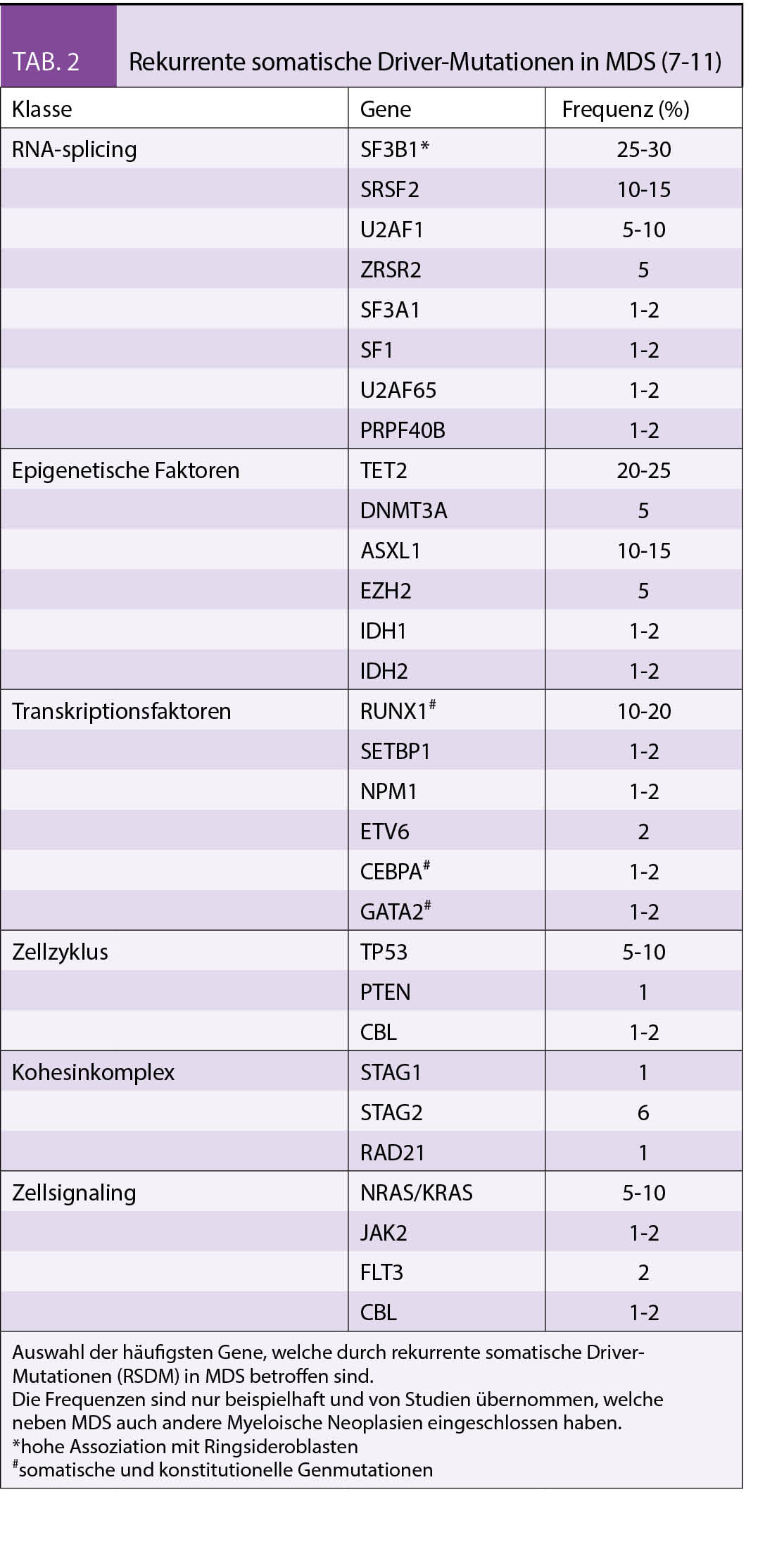
Krankheitsassoziierte Risikofaktoren
Das Risiko für eine Progression in eine sekundäre AML und das Gesamtüberleben kann durch den International Prognostic Scoring System (IPSS) und den revidierten IPSS (IPSS-R) abgeschätzt werden) (22 - 24). Die Anzahl der Zelllinien, die von Zytopenien betroffen sind, wird in allen Prognosescoring-Systemen verwendet. Zudem sind Blastenzahl und Art der zytogenetischen Veränderungen relevant. Weniger als 5% der Blasten im Knochenmark, keine zirkulierenden Blasten im peripheren Blut, isolierte Anämie, Transfusionsunabhängigkeit und normaler Karyotyp oder günstige zytogenetische Veränderungen charakterisieren weniger fortgeschrittene MDS Formen. Im Gegensatz dazu wird ein fortgeschrittenes MDS durch > 5% Blasten im Knochenmark, Zytopenien mehrerer Zelllinien, komplexe zytogenetische oder andere ungünstige Veränderungen definiert.
Patientenassoziierte Risikofaktoren
Bei onkologischen Behandlungen älterer Patienten ist es stets wichtig, die Wirksamkeit und die Verträglichkeit einander gegenüber zu stellen. Karnofsky und Eastern Cooperative Oncology Group (ECOG) Scoring können verwendet werden, um die Performance zu bewerten. Diese sind jedoch altersbedingt und daher nicht ausreichend, um Komorbidität und Gebrechlichkeit zu beurteilen. Der Charlson Komorbiditätsindex wurde von Sorror für Patienten, die sich einer allogenen, hämatopoietischen Stammzelltransplantation (allo-HSCT) unterziehen, angepasst (25, 26) und wurde auch für MDS-Patienten validiert (HCT-CI) (27). Gebrechlichkeit und Funktionalität im täglichen Leben kann mit allgemeinen geriatrischen Bewertungsinstrumenten bewertet werden. Basierend auf einer steigenden Krebsinzidenz bei älteren Patienten sowie einer steigenden Anzahl von zielgerichteten Therapien, ist die Beurteilung patientenbezogener Faktoren ein zunehmendes Erfordernis für eine geeignete Behandlungszuteilung.
MDS-Patienten mit niedrigem Risiko
Watch-and-wait
Die Lebenserwartung von Patienten > 70 Jahre mit MDS-SLD oder MDS mit del(5q) ist nicht signifikant kürzer, als diejenige einer altersangepassten, älteren Bevölkerung (24). Patienten mit niedrig/intermediär-1 IPSS und asymptomatischen Zytopenien sollten daher nur regelmässig kontrolliert werden ohne Behandlung (7). Diese Empfehlung könnte sich in Zukunft jedoch gegebenenfalls ändern, da mit NGS in dieser Patientengruppe auch ungünstige Mutationen nachgewiesen werden können, welche von einer früheren Behandlung profitieren.
Supportive Massnahmen
Die supportiven Massnahmen umfassen Transfusionen, Infektionsprophylaxe, Antiemetika, Analgetika, Eisenchelation und Wachstumsfaktoren. Eine Transfusionsabhängigkeit ist im Allgemeinen mit einem fortgeschrittenen Krankheitsstadium und einer schlechteren Prognose assoziiert (24). Erythrozytenkonzentrate (EKs) werden in der Regel ab einem Hämoglobinspiegel < 80 g / l (oder auf höherem Niveau, falls symptomatisch) transfundiert. Thrombozytenkonzentrate sollten nur zurückhaltend zur Prophylaxe von Blutungen eingesetzt werden. Üblicherweise steigt das Risiko spontaner Blutungen bei Thrombozyten < 5 - 10 G / L oder < 20 G / L mit zusätzlichen Risikofaktoren wie Fieber oder Mukositis. Transfusionen sind in der Regel mit einem höheren Risiko für unerwünschte Ereignisse wie Alloimmunisierung und transfusionsassoziierte Komplikationen assoziiert (28). EKs müssen bei Patienten, die potentielle Kandidaten für eine allo-HSCT sind, bestrahlt werden, um das Risiko einer HLA-Alloimmunisierung und transfusionsassoziierten Graft-versus-Host-Erkrankung zu reduzieren.
Bei stark transfundierten Patienten (> 20 - 25 EKs) und/oder Ferritinwerten > 1000 mg / l kann eine Chelation eine Eisenüberladung vorbeugen, Zytopenien in ca. einem Drittel der Patienten verbessern und möglicherweise auch das Gesamtüberleben günstig beeinflussen (29 - 31). Weiterhin bleibt aber die Frage nicht sicher geklärt, welche Patienten cheliert werden sollen, da bei MDS Patienten (noch) keine randomisierten Studien vorliegen (32). Eine Eisenchelation wird im Allgemeinen mit Deferasirox Patienten angeboten, welche Kandidaten für eine allo-HSCT sind oder eine Lebenserwartung von > 1 Jahr haben.
Patienten mit niedrig/intermediär-1 IPSS, mit Hämoglobinwerten < 100 g / l, Serum-Erythropoietin-Werten < 200 - 500 U/L, EK transfusionsunabhängig oder mit < 2 EKs/Monat sind Kandidaten für eine Behandlung mit Erythropoietin Stimulierenden Agenzien (ESA) (29, 33, 34). Zwischen den verschiedenen ESA-Produkten (rekombinantes humanes Erythropoietin (rHuEPO) Alpha und Beta oder Darbepoietin Alpha) wurden keine relevanten Unterschiede festgestellt. Die erforderlichen EPO-Dosierungen sind höher als bei Patienten mit Niereninsuffizienz und man beginnt in der Regel mit 30 000 U / Woche rHuEPO sc (ca. 150 μg Darbepoietin alpha). Bei fehlendem Ansprechen wird die Dosis nach 6-8 Wochen verdoppelt. Der zusätzliche Einsatz von Granulozyten-Colony Stimulaing Factor (G-CSF 3 x 300 - 480 ug / Woche sc) bei Patienten, die nicht genügend auf ESA ansprechen, ist kontrovers und wird in der Schweiz selten eingesetzt (29, 35, 36). Die Einhaltung eines strikten ESA/G-CSF-Substitutionsregimes ist wichtig, um frühzeitig refraktäre Patienten zu identifizieren, die eine schlechtere Prognose haben und allfällige Kandidaten für weiterführende Behandlungen sein könnten.
Thrombopoietin-stimulierende Agenzien (TSA) (Romiplostim, Eltrombopag) wurden in klinischen Studien bei Patienten mit Thrombozytopenie und niedrig/intermediär-1 IPSS getestet. TSA zeigen eine positive Wirkung auf die Thrombozytenzahl und können Blutungen reduzieren haben aber keinen Einfluss auf das Gesamtüberleben (37, 38). Es ist wichtig zu beachten, dass die Behandlungen mit Wachstumsfaktoren bei MDS-Patienten grundsätzlich zugelassen, aber «off-limitatio» sind und daher eine Kostengutsprache von der zuständigen Krankenkasse notwendig ist.

Krankheitsmodifizierende Behandlungen Immunmodulatorische Medikamente
Die immunmodulatorische und anti-angiogenetische Wirkung von Thalidomid wurde schon früher bei MDS Patienten eingesetzt, um den Transfusionsbedarf zu reduzieren (39). Aufgrund der neurologischen Nebenwirkung von Thalidomid, wurde ein 4-Amino-glutarimid Derivat, das Lenalidomid (Revlimid®) ohne diesen ungünstigen Nebeneffekt entwickelt. Mit Lenalidomid lässt sich eine anhaltende Transfusionsunabhängigkeit und zytogenetische Remission bei etwa der Hälfte aller MDS-Patienten mit niedrig / intermediär-1 IPSS und isoliert del(5q) erreichen (40). In einer Phase-3-Studie fand man zudem auch in ca. einem Viertel der Nicht-del(5q)-MDS-Patienten mit niedrig/intermediär-1 IPSS eine Transfusionsunabhängigkeit, während Mutationen in TP53 mit Resistenz und einer Krankheitsprogression assoziiert waren (41, 42) und daher bei fehlendem Ansprechen Abklärungen in Richtung einer Transplantation rechtfertigen. 10% der MDS-Patienten präsentieren sich mit hypoplastischem Knochenmark und sind potentielle Kandidaten für eine immunsuppressive Behandlung mit Antithymozytenglobulin (ATG) in Kombination mit Cyclosporin A (CyA) mit Ansprechraten von etwa einem Drittel (43). Eine Kombination CyA/ATG mit dem TSA Eltrombopag hat einen zusätzlichen Nutzen bei Patienten mit aplastischer Anämie gezeigt, ist aber für hypoplastische MDS Patienten nicht zugelassen (44).
MDS-Patienten mit höherem Risiko
Hypomethylierende Agenzien
MDS-Patienten mit Blastenexzess oder mit höherem Risiko, welche für eine intensive Chemotherapie und allo-HSCT nicht in Frage kommen, sind Kandidaten für eine palliative Behandlung mit hypomethylierende Agenzien (HMAs). Die Pyrimidin-Nukleosid-Analoga, 5-Azacytidin (AZA) und 5 - Aza - 2’ - desoxycytidin / Decitabine (DEC), wurden in Phase-3-Studien an MDS Patienten mit höherem Risiko untersucht (45,46). HMAs sind im Allgemeinen gut verträglich und zeigten signifikant höhere partielle und vollständige Remissionen im Vergleich zu best supportive care, einschliesslich Hydroxyurea und niedrigdosiertem Cytosin Arabinosid (AraC). HMAs bleiben jedoch der intensivierten Induktions-Chemotherapie gefolgt von allo-HSCT unterlegen, für welche jedoch nur eine Minderheit der älteren MDS Patienten in Frage kommt. MDS mit komplexem Karyotyp, sollten aufgrund der niedrigeren Raten vollständiger Remissionen und der höheren Toxizität mit intensiven Chemotherapien, bevorzugt mit HMA behandelt werden (47). Es gibt keine allgemein anerkannten prädiktiven molekularen Marker für das Ansprechen auf HMA und auch die Dauer der Behandlung bleibt unklar. Derzeit gibt es keine etablierte Behandlung nach Versagen von HMAs, diese Patienten sollten daher vorzugsweise auf klinische Studien behandelt werden.
Intensive Induktionschemotherapie
Die AML-basierte Induktions-Chemotherapie, gefolgt von einer allo-HSCT ist MDS Patienten mit höherem Risiko vorbehalten, die für eine intensive Therapie genügend fit sind. Jüngeres Alter, guter Leistungsstatus und günstige Zytogenetik sind unabhängige prognostische Faktoren, die mit einem besseren Überleben assoziiert sind (48). Patienten mit ungünstigen oder komplexen zytogenetischen Veränderungen sowie Mutationen oder Deletionen in TP53 haben ein schlechteres Ansprechen auf eine intensive Chemotherapie und können von einer Behandlung mit HMA mit oder ohne anschliessender allo-HSCT profitieren (47, 49). Die derzeitige Datenlage ist aber im Allgemeinen noch nicht ausreichend, um HMA für die Induktion vor allo-HSCT ausserhalb klinischer Studien zu empfehlen (8).
Allogene hämatopoietische Stammzelltransplantation (allo HSCT)
Die allo HSCT bleibt die einzige kurative Option, ist aber nur für MDS-Patienten geeignet, die genügend fit sind. Die Beurteilung der Komorbiditäten ist wichtig für die Entscheidungsfindung, welche Patienten für eine allo-HSCT in Frage kommen und der HCT-CI Score wird oft für diesen Zweck verwendet (26). Für eine allo HSCT kommen MDS Patienten mit intermediär-2/hoch IPSS in Frage. Das Alter ist der wichtigste prädiktive Faktor für das Gesamtüberleben. Eine retrospektive Analyse der Europäischen Gruppe für Blut- und Knochenmarktransplantation (EBMT) ergab eine behandlungsassoziierte Mortalität von 30% bei Patienten < 20 Jahre, 43% bei Patienten zwischen 20 und 40 Jahren und 50% bei Patienten> 50 Jahre. Reduktion der Intensität der Konditionierung und sorgfältige Auswahl der Patienten haben jedoch gezeigt, dass eine allo-HSCT auch bei Patienten zwischen 60 und 70 Jahren möglich ist. Für MDS-Patienten, die aufgrund von Komorbiditäten nicht für eine myeloablative Konditionierung qualifizieren, kann eine Konditionierung mit reduzierter Intensität in Betracht gezogen werden, vorzugsweise innerhalb klinischer Studien.
Zukunftsperspektiven
Die Entdeckung von RSDM bei Patienten mit myeloischen Neoplasien eröffnet eine ungeahnte Palette neuer diagnostischer und therapeutischer Möglichkeiten. Eine klonale Evolution kann damit bereits in frühen Stadien einer klonalen Hämatopoese indentifiziert werden und erlaubt möglicherweise auch die Identifikation von Patienten, welche von einer früheren Intervention profitieren können. Es konnte zum Beispiel gezeigt werden, dass Mutationen in TP53, EZH2, ETV6, RUNX1 und ASXL1 mit einer schlechten Prognose assoziiert sind und ein molekulares Scoring-System wird derzeit entwickelt (IPSS-R Mole) (50). Bei MDS-Patienten mit niedrigem Risiko und RS- oder SF3B1-Mutationen (MDS-RS-SLD / MLS) führt der TGF-beta ligand-trap Luspatercept in zwei Drittel der Patienten zu einem erythroiden Ansprechen und Transfusionsfreiheit. Luspatercept scheint die Erythropoese durch Mechanismen zu verbessern, die unabhängig von EPO sind. Basierend auf der Annahme, dass eine niedrig dosierte und längere Exposition mit HMAs zu einer Verbesserung der Differenzierung führen kann, werden zur Zeit orale AZA-Formulierungen in klinischen Studien an MDS-Patienten mit niedrigem Risiko getestet (51). Weitere Medikamente, die an MDS Patienten untersucht werden, umfassen Toll-like-Rezeptor-2-Antikörper, CD95-Ligand(FAS-Ligand)-Hemmer, Multikinase-Inhibitoren (z. B. Rigosertib), Checkpoint-Inhibitoren (z. B. Nivolumab, Durvalumab), Telomerase-Inhibitoren (z. B. Imetelstat) und Inhibitoren von mutiertem IDH1 / 2 (zB AG - 120 / AG-221).
Darüber hinaus ist die Verbesserung der Behandlungsallokation basierend auf Wirksamkeit, Verträglichkeit, Nutzen und Richtlinien-Konformität ein aktives Forschungsgebiet der Versorgungsforschung. Daher ist der Einschluss von MDS-Patienten in longitudinale Kohortenstudien sehr zu begrüssen, so wie es die SAKK 33/18 («I-CARE for MDS») Studie verfolgen wird.
Weiter werden longitudinale Kohorten und Biobanking von biologischem Material von Patienten mit MDS oder frühen Formen einer klonalen Hämatopoese (CCUS) sehr nützlich sein, um detaillierte, gesundheitsbezogene Daten mit Biomarkern zu verknüpfen. Diese Plattform steht seit 2016 mit dem Swiss MDS Regsitry/Biobank zur Verfügung und soll in Zukunft helfen, unser Verständnis der MDS Biologie zu verbessern, um damit auch im Sinne einer «Präzisionsmedizin» die Prognose und das Therapieansprechen der Patienten besser abschätzen zu können.
Zusammenfassung
Die Myelodysplastischen Syndrome (MDS) bilden eine heterogene Gruppe von klonalen Erkrankungen des Blutes mit einer zunehmenden Inzidenz in der älteren Bevölkerung. Aufgrund der demographischen Alterung unserer Gesellschaft werden MDS eine wachsende Bedeutung in unserem Gesundheitswesen haben. MDS werden durch Genmutationen in den hämatopoetischen Stammzellen verursacht und sind gekennzeichnet durch eine ineffektive Hämatopoiese mit Zytopenien und Dysplasien sowie einer Neigung zur Progression in eine sekundäre akute myeloische Leukämie (sAML). Eine exakte Diagnose und Risikostratifizierung sind für eine korrekte Behandlung entscheidend. Patienten mit niedrigem Risiko haben ein medianes Überleben von 3 bis 8 Jahren und sterben vorwiegend an zytopenieassoziierten Komplikationen (kardiovaskuläre Ereignisse, Infektionen und Blutungen). Die Ziele einer Behandlung von Patienten mit niedrigem Risiko liegen daher vor allem in der Verbesserung der Lebensqualität, Verringerung von zytopenieassoziierten Komplikationen und einem Hinauszögern eines Progresses in ein höhergradiges MDS. Bei MDS Patienten mit hohem Risiko liegt hingegen die mediane Überlebenszeit nur bei 1 bis 3 Jahren und die AML-assoziierte Mortalität steht im Vordergrund. Bei diesen Patienten sollte die Behandlung darauf ausgerichtet sein, die Progression in eine AML hinauszuzögern und das Gesamtüberleben zu verbessern. Die allogene hämatopoetische Stammzelltransplantation bleibt die einzige kurative Option für Patienten mit hohem Risiko. Jedoch ist nur eine Minderheit der meistens älteren und polymorbiden MDS Patienten geeignet für eine solche intensive Behandlung. Daher werden die meisten Patienten mit supportiven Massnahmen und palliativen Behandlungen, wie zum Beispiel Wachstumsfaktoren, Immunmodulatoren und hypomethylierenden Agenzien behandelt. Da ältere Patienten mit chronischen Zytopenien häufig in der allgemein internistischen Praxis gesehen werden, ist die Kenntnis über mögliche Präsentationsformen und angemessene Behandlungsoptionen wichtig für alle Ärzte aus der Grundversorgung.
Leitender Arzt und Koordinator Swiss MDS Study Group
Universitätsklinik für Hämatologie und Hämatologisches Zentrallabor
Inselspital Bern
Freiburgstrasse 18
3010 Bern
Nicolas.Bonadies@insel.ch
Der Autor hat deklariert, keine Interessenskonflikte in Zusammenhang mit diesem Artikel zu haben.
1. Bonadies N, Feller A, Rovo A, et al. Trends of classification, incidence, mortality, and survival of MDS patients in Switzerland between 2001 and 2012. Cancer Epidemiol 2017;46:85-92.
2. Dinmohamed AG, Visser O, van Norden Y, et al. Trends in incidence, initial treatment and survival of myelodysplastic syndromes: a population-based study of 5144 patients diagnosed in the Netherlands from 2001 to 2010. Eur J Cancer 2014;50:1004-12.
3. Roman E, Smith A, Appleton S, et al. Myeloid malignancies in the real-world: Occurrence, progression and survival in the UK’s population-based Haematological Malignancy Research Network 2004-15. Cancer Epidemiol 2016;42:186-98.
4. Neukirchen J, Schoonen WM, Strupp C, et al. Incidence and prevalence of myelodysplastic syndromes: data from the Dusseldorf MDS-registry. Leuk Res 2011;35:1591-6.
5. Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer 2007;7:118-29.
6. Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:504-15.
7. Malcovati L, Hellstrom-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 2013;122:2943-64.
8. Abdel-Wahab O, Figueroa ME. Interpreting new molecular genetics in myelodysplastic syndromes. Hematology American Society of Hematology Education Program 2012;2012:56-64.
9. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011;364:2496-506.
10. Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011;478:64-9.
11. Kon A, Shih LY, Minamino M, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 2013;45:1232-7.
12. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391-405.
13. Valent P, Horny HP. Minimal diagnostic criteria for myelodysplastic syndromes and separation from ICUS and IDUS: update and open questions. Eur J Clin Invest 2009;39:548-53.
14. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477-87.
15. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488-98.
16. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9-16.
17. Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001-2004, using data from the NAACCR and SEER programs. Blood 2008;112:45-52.
18. Santini V. Treatment of low-risk myelodysplastic syndromes. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program 2016;2016:462-9.
19. Fenaux P, Ades L. How we treat lower-risk myelodysplastic syndromes. Blood 2013;121:4280-6.
20. Komrokji RS. Current State of the Art: Management of Higher Risk Myelodysplastic Syndromes. Clinical lymphoma, myeloma & leukemia 2016;16 Suppl:S39-43.
21. Garcia-Manero G. Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management. American journal of hematology 2015;90:831-41.
22. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997;89:2079-88.
23. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012;120:2454-65.
24. Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2007;25:3503-10.
25. Charlson ME, Pompei P, Ales KL, MacKenzie CR. A new method of classifying prognostic comorbidity in longitudinal studies: development and validation. J Chronic Dis 1987;40:373-83.
26. Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 2005;106:2912-9.
27. Sorror ML, Storb RF, Sandmaier BM, et al. Comorbidity-age index: a clinical measure of biologic age before allogeneic hematopoietic cell transplantation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2014;32:3249-56.
28. Szczepiorkowski ZM, Dunbar NM. Transfusion guidelines: when to transfuse. Hematology American Society of Hematology Education Program 2013;2013:638-44.
29. Malcovati L, Hellstrom-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 2013;122:2943-64.
30. Gattermann N, Finelli C, Della Porta M, et al. Hematologic responses to deferasirox therapy in transfusion-dependent patients with myelodysplastic syndromes. Haematologica 2012;97:1364-71.
31. List AF, Baer MR, Steensma DP, et al. Deferasirox reduces serum ferritin and labile plasma iron in RBC transfusion-dependent patients with myelodysplastic syndrome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30:2134-9.
32. Mitchell M, Gore SD, Zeidan AM. Iron chelation therapy in myelodysplastic syndromes: where do we stand? Expert Rev Hematol 2013;6:397-410.
33. Park S, Grabar S, Kelaidi C, et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood 2008;111:574-82.
34. Hellstrom-Lindberg E, Negrin R, Stein R, et al. Erythroid response to treatment with G-CSF plus erythropoietin for the anaemia of patients with myelodysplastic syndromes: proposal for a predictive model. British journal of haematology 1997;99:344-51.
35. Balleari E, Rossi E, Clavio M, et al. Erythropoietin plus granulocyte colony-stimulating factor is better than erythropoietin alone to treat anemia in low-risk myelodysplastic syndromes: results from a randomized single-centre study. Ann Hematol 2006;85:174-80.
36. Casadevall N, Durieux P, Dubois S, et al. Health, economic, and quality-of-life effects of erythropoietin and granulocyte colony-stimulating factor for the treatment of myelodysplastic syndromes: a randomized, controlled trial. Blood 2004;104:321-7.
37. Kantarjian H, Fenaux P, Sekeres MA, et al. Safety and efficacy of romiplostim in patients with lower-risk myelodysplastic syndrome and thrombocytopenia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2010;28:437-44.
38. Mavroudi I, Pyrovolaki K, Pavlaki K, et al. Effect of the nonpeptide thrombopoietin receptor agonist eltrombopag on megakaryopoiesis of patients with lower risk myelodysplastic syndrome. Leuk Res 2011;35:323-8.
39. Raza A, Meyer P, Dutt D, et al. Thalidomide produces transfusion independence in long-standing refractory anemias of patients with myelodysplastic syndromes. Blood 2001;98:958-65.
40. Fenaux P, Giagounidis A, Selleslag D, et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011;118:3765-76.
41. Santini V, Almeida A, Giagounidis A, et al. Randomized Phase III Study of Lenalidomide Versus Placebo in RBC Transfusion-Dependent Patients With Lower-Risk Non-del(5q) Myelodysplastic Syndromes and Ineligible for or Refractory to Erythropoiesis-Stimulating Agents. J Clin Oncol 2016;34:2988-96.
42. Jadersten M, Saft L, Smith A, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:1971-9.
43. Passweg JR, Giagounidis AA, Simcock M, et al. Immunosuppressive therapy for patients with myelodysplastic syndrome: a prospective randomized multicenter phase III trial comparing antithymocyte globulin plus cyclosporine with best supportive care–SAKK 33/99. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:303-9.
44. Olnes MJ, Scheinberg P, Calvo KR, et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med 2012;367:11-9.
45. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009;10:223-32.
46. Lubbert M, Suciu S, Baila L, et al. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011;29:1987-96.
47. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015;126:291-9.
48. Ossenkoppele GJ, Graveland WJ, Sonneveld P, et al. The value of fludarabine in addition to ARA-C and G-CSF in the treatment of patients with high-risk myelodysplastic syndromes and AML in elderly patients. Blood 2004;103:2908-13.
49. Knipp S, Hildebrand B, Kundgen A, et al. Intensive chemotherapy is not recommended for patients aged >60 years who have myelodysplastic syndromes or acute myeloid leukemia with high-risk karyotypes. Cancer 2007;110:345-52.
50. Nazha A, Narkhede M, Radivoyevitch T, et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 2016.
51. Saunthararajah Y. Key clinical observations after 5-azacytidine and decitabine treatment of myelodysplastic syndromes suggest practical solutions for better outcomes. Hematology American Society of Hematology Education Program 2013;2013:511-21.

info@onco-suisse
- Vol. 8
- Ausgabe 4
- August 2018