2021 liess Swissmedic 45 Humanarzneimittel mit neuen Wirkstoffen zu
Über alle Verfahren gepoolt betrug die Durchlaufzeit der Gesuche im Median 396 Kalendertage (Vorjahr: 482 Kalendertage). Damit liegt Swissmedic erstmals unter dem mehrjährigen Mittel der EMA von ca. 400 Tagen.
- In 7% der Fälle kam das beschleunigte Zulassungsverfahren zur Anwendung. Die Durchlaufzeit der Gesuche betrug in diesem Verfahren im Median 207 Kalendertage.
- Das Verfahren mit Voranmeldung mit um 20% verkürzter Swissmedic- Begutachtungszeit kam in einem Fall zur Anwendung. Die Durchlaufzeit betrug in diesem Verfahren 305 Kalendertage.
- Die befristeten Zulassungen machen 24% der Arzneimittel aus: Von den 11 befristeten Zulassungen wurden 5 Arzneimittel von Firmen befristet beantragt und entsprechend beschleunigt begutachtet. Die Durchlaufzeit dieser Gesuche betrug im Median 264 Kalendertage. 6 weitere Gesuche wurden von Swissmedic von Amtes wegen befristet zugelassen. Dazu gehörten die beiden COVID-19-Impfstoffe Spikevax (61 Kalendertage) und COVID-19 Vaccine Janssen (109 Kalendertage).
- In 22% der Fälle kamen die vereinfachten Zulassungsverfahren nach Art. 13 und Art. 14 Abs. 1 Bst. abis-quater HMG zur Anwendung.
- 18% der innovativen Neuzulassungen erfolgten im Rahmen von internationalen Verfahren: Im Geschäftsjahr wurden 5 Arzneimittel im Rahmen des Projektes Orbis und 3 Arzneimittel im Worksharing-Verfahren des Access Consortiums begutachtet.
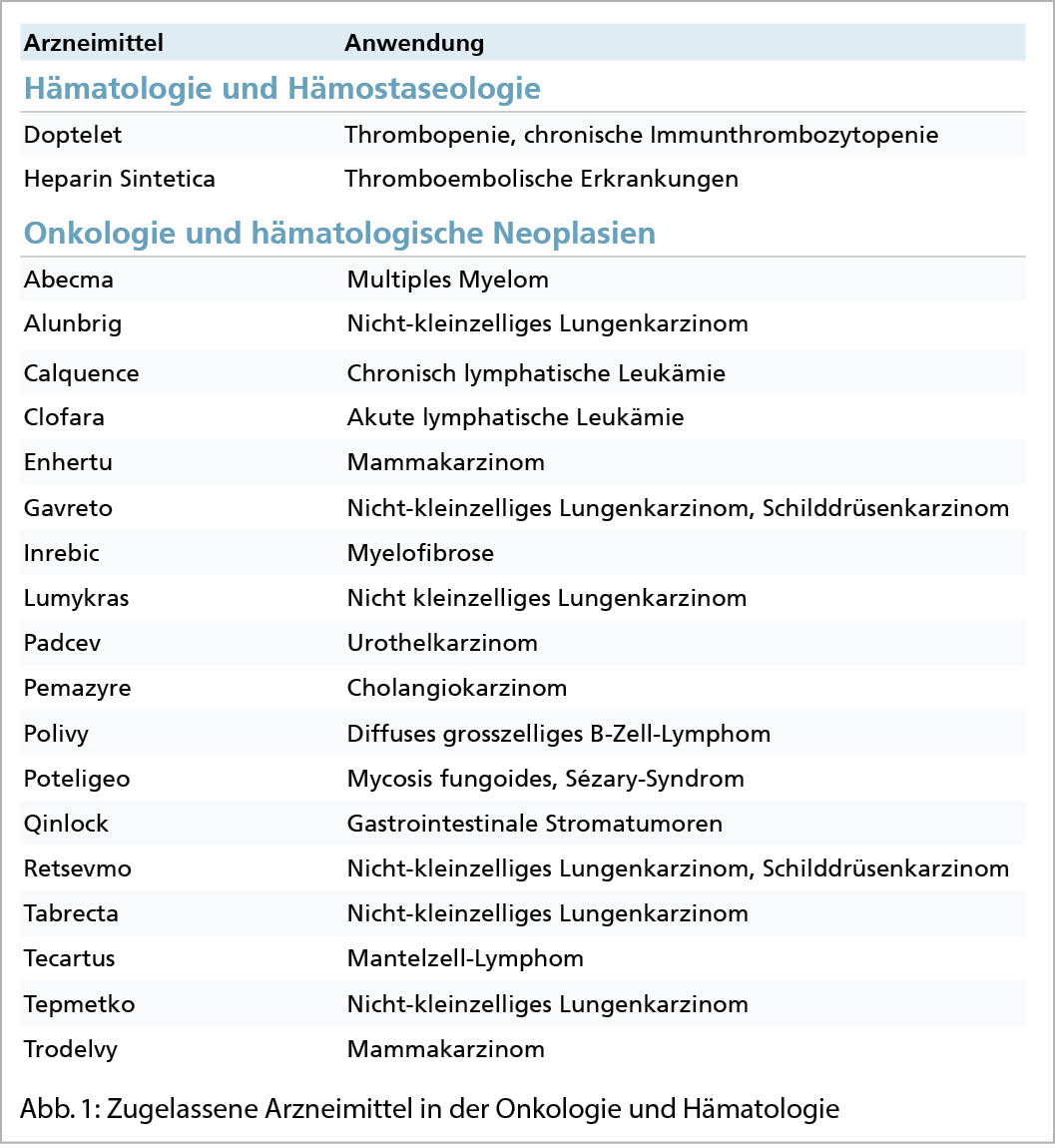
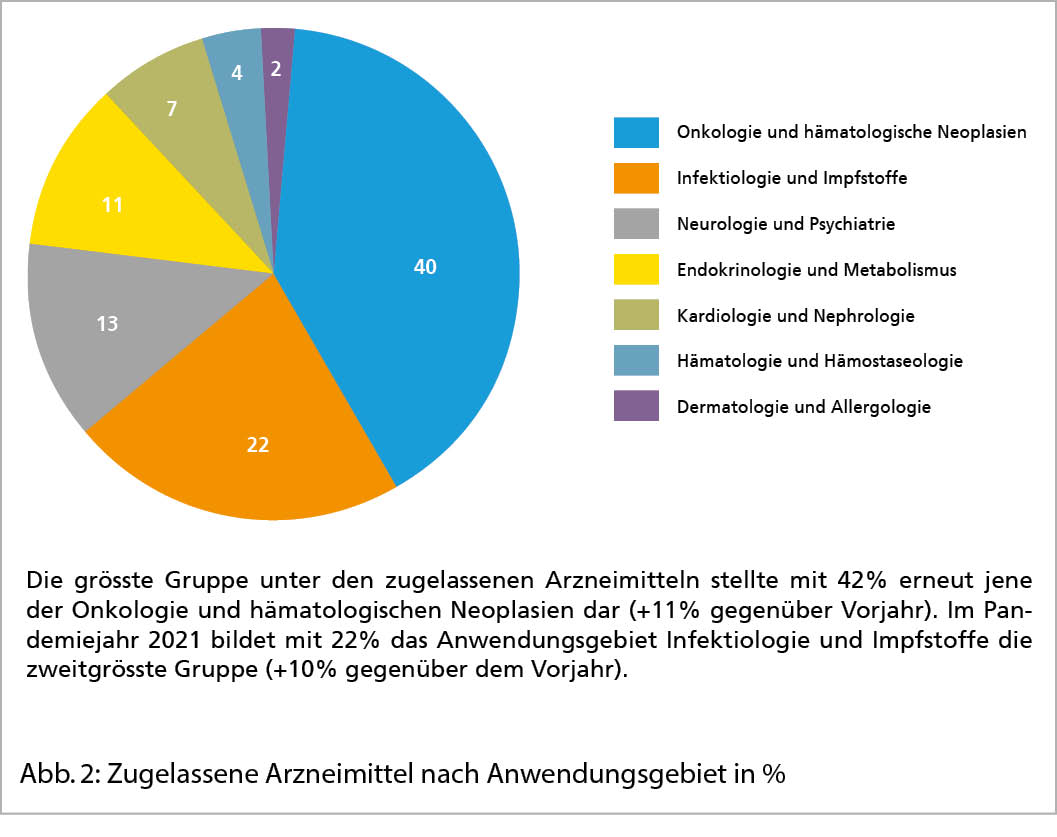
2021 liess Swissmedic 84 Indikationserweiterungen zu
Über alle Verfahren gepoolt betrug die Durchlaufzeit der Gesuche im Median 348 Kalendertage.
- In 2 Fällen kam das beschleunigte Zulassungsverfahren zur Anwendung. Die Durchlaufzeit betrug im Median 214 Kalendertage.
- Das Verfahren mit Voranmeldung (VmVA) mit um 20% verkürzter Swissmedic-Begutachtungszeit kam in 8% der Fälle zur Anwendung. Indikationserweiterungen im VmVA konnten 2021 im Median nach nur 302 Kalenderta¬gen abgeschlossen werden.
- In 12% der Fälle kamen die vereinfachten Zulassungsverfahren nach Art. 13 und Art. 14 Abs. 1 Bst. abis-quater HMG zur Anwendung.
- Im Geschäftsjahr wurden zudem 7 Indikationserweiterungen im Rahmen des Projektes Orbis zugelassen. Diese Gesuche konnten im Median nach 216 Kalendertagen abgeschlossen werden.
*Projekt Orbis: Das Oncology Center of Excellence (OCE) der U.S. Food and Drug Administration (FDA) hat im September 2019 unter dem Namen «Orbis» ein Projekt initiiert, um den Zulassungsprozess neuer Krebstherapien weltweit zu beschleunigen. Pharmazeutische Firmen können im Rahmen des Projekts Orbis ihre Zulassungsgesuche neben der FDA simultan bei weiteren teilnehmenden internationalen Zulassungsbehörden einreichen. Die entsprechenden Gesuche werden von diesen Zulassungsbehörden in Zusammenarbeit mit der FDA parallel geprüft und eine allfällige Zulassung dadurch beschleunigt. Damit erhalten auch Schweizer Krebspatientinnen und -patienten rascher Zugang zu innovativen Therapiemöglichkeiten.
Swissmedic unterstützt das Projekt «Orbis» in Übereinstimmung mit ihren strategischen Zielen und nahm 2020 im Rahmen eines Piloten am Projekt teil. Nach Evaluation der gewonnenen Erfahrung hat Swissmedic nun entschieden, das Projekt «Orbis» permanent weiterzuführen. www.swissmedic.ch
Quelle: Swissmedic Newsletter 01.03.2022
DHPC – Alecensa® (Alectinib)
Warnhinweise und Vorsichtsmassnahmen sowie spezifische Anleitung zur Dosisänderung im Falle einer hämolytischen Anämie
Roche Pharma (Schweiz) AG informierte in Absprache mit Swissmedic über folgenden Sachverhalt:
- In klinischen Studien und nach der Markteinführung wurden Fälle von hämolytischer Anämie berichtet, die als Risiko einer Therapie mit Alecensa zu betrachten sind.
- Eine kürzlich durchgeführte kumulative Analyse von Fällen mit hämolytischer Anämie zeigte, dass eine Dosisänderung von Alecensa in der Mehrheit der Fälle eine Verbesserung der hämolytischen Anämie bewirkte.
- Die Behandlung mit Alecensa sollte zunächst unterbrochen und geeignete Labortests sollten eingeleitet werden, wenn die Hämoglobinkonzentration < 10 g/dl beträgt und eine hämolytische Anämie vermutet wird.
- Wird eine hämolytische Anämie bestätigt, so sollte die Behandlung mit Alecensa bis zum Abklingen des Ereignisses unterbrochen und mit einer reduzierten Dosis wieder aufgenommen oder definitiv beendet werden. Das Vorgehen bei der Dosisreduktion wird im Abschnitt Dosierung/Anwendung der Fachinformation beschrieben.
Quelle: Swissmedic, 15.02.2022
Padcev® (Wirkstoff: Enfortumab vedotin)
Erstzulassung in der Schweiz: 09.11.2021 Arzneimittel zur Behandlung von Urothelkarzinomen bei Erwachsenen
Über das Arzneimittel
Padcev enthält den Wirkstoff Enfortumab vedotin und wird angewendet zur Behandlung von Erwachsenen mit Urothelkarzinom (mUC) (1), welches lokal fortgeschritten oder metastasiert ist.
Patientinnen und Patienten, die für diese Behandlung in Frage kommen, hatten zuvor bereits eine platinhaltige Chemotherapie und wurden während bzw. nach der Therapie mit Immuncheckpointinhibitoren (PD-1/PD-L1 (2)) behandelt und haben ein Fortschreiten oder einen Rückfall der Krankheit erlitten.
Wirkung
Enfortumab vedotin gehört zur Medikamentenklasse der Antikörper-Arzneimittel-Konjugate (ADC). Der Wirkstoff besteht aus einem monoklonalen Antikörper (immunologisch aktive Proteine), der mit der Substanz Monomethyl-Auristatin E (MMAE) verbun-den ist. MMAE ist ein Cytotoxin (Zellgift), das Krebszellen abtöten kann. Der monoklonale Antikörper bindet sich vorwiegend an einen spezifischen Rezeptor (Zielstelle) an der Oberfläche der Urothelkarzinomzellen, wodurch MMAE in die Zellen freigesetzt wird. Der damit ausgelöste Prozess führt zum Zelltod der Krebszelle.
Anwendung
Padcev ist rezeptpflichtig und als Pulver für ein Konzentrat zur Herstellung einer Infusionslösung zugelassen. Es wird als Flüssigkeit in die Venen verabreicht. Die Durchstechflaschen enthalten 20 mg bzw. 30 mg Enfortumab vedotin. Die empfohlene Dosis beträgt 1,25 mg/kg Körpergewicht bis zu 125 mg und es wird über 30 Minuten an den Tagen 1, 8 und 15 eines 28-tägigen Zyklus verabreicht bis zum Fortschreiten der Krankheit oder inakzeptablen Nebenwirkungen.
Wirksamkeit
Die Wirksamkeit von Padcev zur Behandlung von Urothelkarzinomen wurde vor allem in der Studie EV-301 mit insgesamt 608 Teilnehmenden untersucht. Die Patientinnen und Patienten hatten ein lokal fortgeschrittenes oder metastasiertes Urothelkarzinom, waren zuvor mit einer platinhaltigen Chemotherapie behandelt worden und hatten während oder nach der Therapie mit Immuncheckpointinhibitoren (PD-1/PD-L1 Inhibitor) einen Rückfall oder ein Fortschreiten der Erkrankung erlitten. Um die Wirksamkeit von Padcev zu bestätigen, wurde die Hälfte der Patientinnen und Patienten mit Padcev behandelt und die andere Hälfte mit einer Chemotherapie, welche vom Prüfarzt festgelegt wurde.
Die Studie zeigte eine statistisch signifikante Verbesserung des Gesamtüberlebens (3) sowie des progressionsfreies Überleben (4) und der objektiven Ansprechrate (5) der Patientinnen und Patienten, die mit Padcev behandelt wurden, im Vergleich zu jenen, die eine Chemotherapie erhielten.
Vorsichtsmassnahmen, unerwünschte Wirkungen & Risiken
Padcev darf bei einer Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe nicht angewendet werden. Die häufigsten unerwünschten Wirkungen aller mit Padcev behandelten Patientinnen und Patienten waren Alopezie (übermässiger Haarausfall), Müdigkeit, verminderter Appetit, periphere sensorische Neuropathie (Erkrankung des Nervensystems), Durchfall, Übelkeit, Pruritus (Juckreiz), Dysgeusie (Geschmacksstörung), Anämie (Blutarmut), Gewichtsabnahme, makulopapulöser (knotig-fleckiger) Ausschlag, trockene Haut, Erbrechen, erhöhte AST/ ALT (6), Hyperglykämie (zu hoher Blutzucker), trockenes Auge und Ausschlag. Padcev kann andere schwerwiegende Nebenwirkungen verursachen über die der Arzt bzw. die Ärztin unverzüglich in Kenntnis gesetzt werden muss (z. B. schwere Hautnebenwirkungen, akute Nierenverletzung, Lungenentzündung, Harnwegsinfek-tion und Sepsis). Alle Vorsichtsmassnahmen, Risiken und weitere mögliche unerwünschte Wirkungen werden in der Fachinformation aufgeführt.
Begründung des Zulassungsentscheids
Patientinnen und Patienten mit Urothelkarzinom, bei denen die Krankheit nach einer platinbasierten Chemotherapie und an-schliessender Therapie mit Immuncheckpointinhibitoren (PD-1/ PD-L1 Inhibitor) fortschreitet, haben eine schlechte Prognose und es stehen nur begrenzt weitere Behandlungsmöglichkeiten zur Verfügung. Die zulassungsrelevante Studie zeigte einen statistisch signifikanten und klinisch bedeutsamen Vorteil von Padcev im Vergleich zur Kontrollgruppe mit einer Verlängerung der medianen (7) Gesamtüberlebenszeit um 3,9 Monate Unter Berücksichtigung aller vorliegenden Daten überwiegen die Vorteile von Padcev die Risiken. Swissmedic hat daher das Arzneimittel Padcev zugelassen für die Behandlung von Erwachsenen mit Urothelkarzinom (mUC), welches lokal fortgeschritten oder metastasiert ist, wenn zuvor bereits eine platinhaltige Chemotherapie Behandlung stattgefunden hat und während bzw. nach der Therapie mit Immunchekpointinhibotoren (PD-1/ PD-L1) ein Fortschreiten oder einen Rückfall der Krankheit erlitten wurde.
Quelle: Public Summary SwissPAR vom 09.03.2022