Sarkome sind seltene Tumore: die altersstandardisierte Erkrankungsrate in der Schweiz lag 2011 – 2015 bei 4.43/100 000/Jahr für Weichgewebssarkome und bei 0.91/100 000/Jahr für Knochensarkome (1). Die aktuelle 5. Edition der WHO Klassifikation listet über 100 Entitäten intermediärer (lokal aggressiv, sehr selten metastasierend) und maligner Weichgewebs- und Knochentumore. Entsprechend ist die Inzidenz auch bei den häufigeren Entitäten wie dem Liposarkom und dem Leiomyosarkom < 1/100 000/Jahr.
Um Fortschritte in der Behandlung von Sarkomen zu erzielen, ist – wie bei allen seltenen Erkrankungen – die internationale Zusammenarbeit in der Forschung und ein Wissenstransfer zentral, ebenso die Behandlung der Patienten durch erfahrene multidisziplinäre Teams. Dies bedingt den Aufbau von Referenzzentren respektive Referenznetzwerken sowie die entsprechende Zuweisung von Patienten. Bei den Sarkomerkrankungen relevant ist, dass die Zuweisung bereits zum Zeitpunkt der bildgebenden Verdachtsdiagnose und noch vor der Biopsie erfolgt, dies setzt das entsprechende Wissen um Abklärungsalgorithmen beim Primärbehandler voraus. Diagnostik und Behandlung in Referenzzentren korreliert positiv mit dem Gesamtüberleben der Patienten und hat entsprechend Eingang in die Guidelines gefunden (2, 3).
Fortschritte sind im letzten Jahr in verschiedenen Bereichen zu verzeichnen, worauf in der Folge eingegangen werden soll.
WHO Klassifikation 5. Edition
Seit diesem Jahr gilt die 5. Edition der WHO-Klassifikation der Weichgewebs- und Knochentumore. Die Fortschritte in der molekularen Charakterisierung verschiedener Sarkomentitäten finden darin Eingang. So wurde die Gruppe der ehemals Ewing-like Sarkome in die rundzelligen Sarkome mit EWSR1-non-ETS Fusionen, CIC-rearrangierte Sarkome und Sarkome mit BCOR Alteration aufgeteilt. Dies bei klinisch differentem, meist aggressiverem Verlauf verglichen mit dem klassischen Ewing Sarkom (4). Ebenso wurden neu die Gruppe der NTRK-rearranged spindel cell neoplasm und der EWSR1-SMAD3-positive fibroblastic tumors geschaffen. Nicht immer ist die molekulare Neuzuordnung bislang mit therapeutischen Konsequenzen verbunden. Die häufiger durchgeführten molekularen Analysen haben zudem die Anzahl bekannter Translokationen deutlich erhöht – dabei ist die Frage offen, wann diese Genfusionen auch Driver sind und damit in Zukunft therapeutisch angegangen werden können (5)
Ewing Sarkom
Am ASCO 2020 wurden die Resultate der Euro Ewing 2012 / R1-Randomisierung (6), der Ewing 2008 / R3-Arm (9) sowie die zweite Interimsanalyse der rEECur Studie (12) präsentiert.
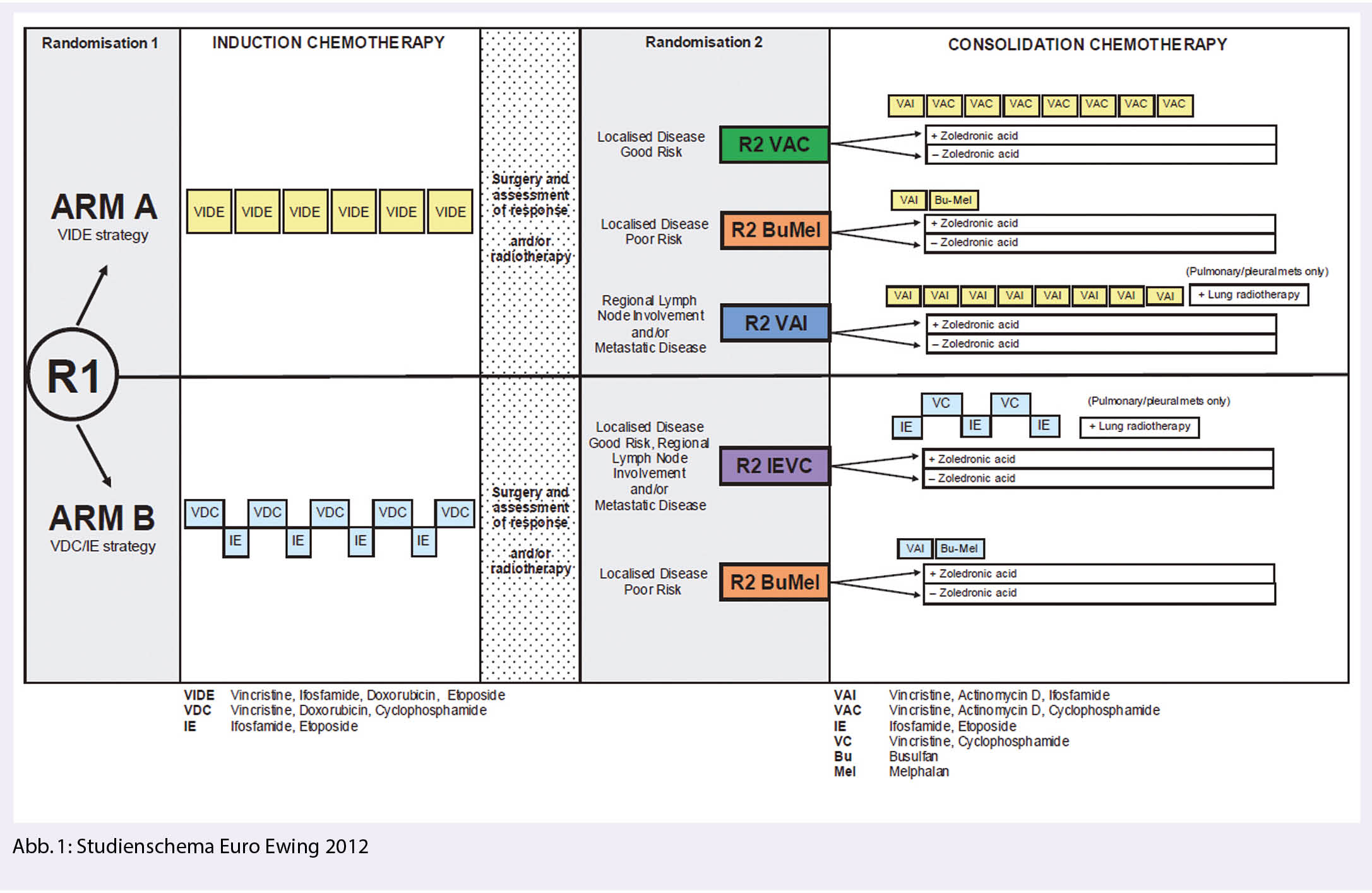
Euro Ewing 2012: Annäherung in der Definition eines internationalen Standards für die Erstlinien Behandlung des Ewing Sarkoms
Die R1 Randomisierung der Euro Ewing 2012 verglich den europäische Behandlungsstandard (Arm A: Induktion mit 6 Zyklen VIDE, Lokaltherapie, Konsolidierung mit 8 Zyklen VAC/VAI respektive bei lokalisierter Erkrankung mit hohem Risiko 1 Zyklus VAI/Hochdosischemotherapie mit Busulfan/Melphalan) mit der amerikanischen Standardbehandlung (Arm B: Induktion mit VDC/IE 2-wöchentlich, Lokaltherapie, Konsolidierung mit IE/VC 2-wöchentlich), wobei Patienten mit poor risk lokalisierter Erkrankung im Arm B analog dem europäischen Standard mit Hochdosischemotherapie behandelt wurden (Abb. 1). In der Schweiz haben die Zentren Bern, Zürich und St.Gallen an der Studie teilgenommen.
Der Arm B zeigte sich sowohl bezüglich Ereignis-freiem Überleben (event free survival, EFS) wie bezüglich Gesamtüberleben (overall survival, OS) klar überlegen, sodass die R1 Randomisierung nach Erreichen der statistisch geforderten Minimalanzahl Patienten im Mai 2019 geschlossen wurde. Die nun gezeigten Daten bestätigen mit einer Hazard Ratio (HR) von 0.70 (95% CI 0.51- 0.95) für das EFS und einer HR von 0.64 (95% CI 0.42 -0.96) für das OS den Vorteil des komprimierten VDC/IE Schemas. Die Toxizitäten waren vergleichbar (Arm A 68% serious adverse events (SAE) vs. Arm B 67% SAEs) (6). Somit wird der europäische Standard in der Erstlinientherapie des Ewing Sarkoms durch die amerikanische Standardbehandlung abgelöst, ergänzt durch eine Hochdosischemotherapie bei lokalisierter Erkrankung mit hohem Risiko.
Diese Resultate bestätigen den Benefit einer dosisintensiveren Behandlung beim Ewing Sarkom, wie dies bereits vom Benefit der Hochdosischemotherapie bekannt ist. In welchem Ausmass damit vermehrt Langzeittoxizitäten in Kauf genommen werden müssen wird sich weisen, sicherlich sind diesbezüglich gewisse Limiten erreicht. Dringend sollten auch deswegen neue Substanzen in die Behandlung des Ewing Sarkoms integriert werden können. Die Voraussetzung einer einheitlichen Erstlinienbehandlung für zukünftige Studien in möglichst interkontinentaler Zusammenarbeit ist hiermit nun gegeben (7, 8).
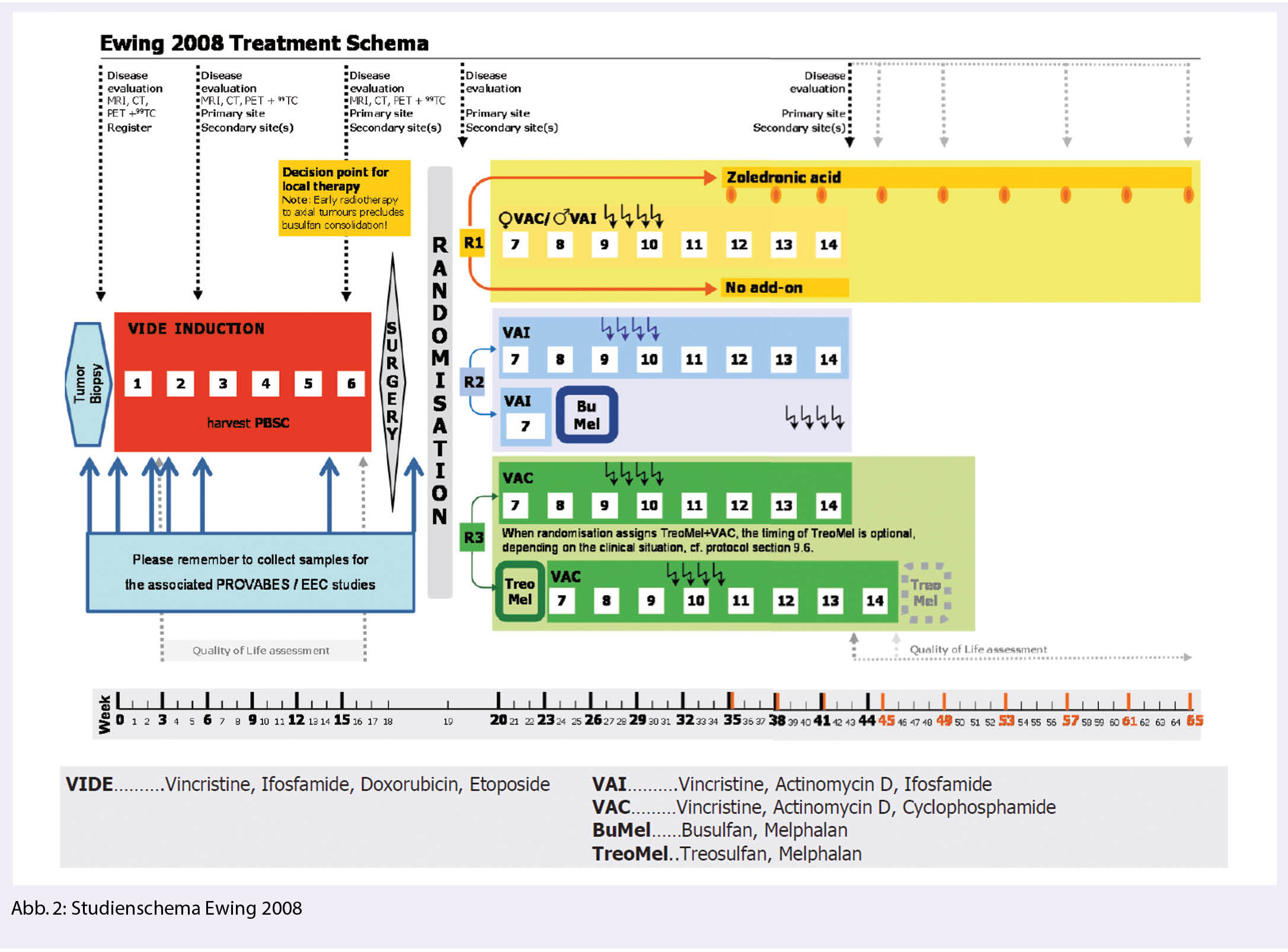
Hochdosischemotherapie beim Ewing Sarkom: indiziert ausschliesslich bei lokalisierter Erkrankung mit hohem Risiko
Die Ewing 2008 Studie untersuchte in ihrem dritten Studienarm bei Patienten mit primär disseminierter Erkrankung den Nutzen einer zusätzlichen Hochdosischemotherapie mit Treosulfan und Melphan (Abb. 2). Das 3-Jahres EFS war mit und ohne Hochdosistherapie in der Gesamtkohorte vergleichbar bei 20.9% (95% CI 11.5 – 37.9%) vs. 19.2% (95% CI 10.8 – 34.4.%). Nur für Patienten < 14 Jahre zeigte sich ein möglicher Vorteil der Hochdosischemotherapie (3 Jahre EFS 39.9% vs. 9%) (9).
Der Stellenwert der Hochdosischemotherapie in der Erstlinienbehandlung des Ewing Sarkoms nach Induktionstherapie mit VIDE hat sich somit geklärt und ist einzig bei Patienten mit lokalisierter Hochrisiko Erkrankung gegeben. Eine lokalisierte Erkrankung wird der Hochrisikogruppe zugeordnet, wenn sich nach Induktionschemotherapie ein schlechtes pathologisches Ansprechen zeigt (≥ 10% residuelle Tumorzellen) respektive initial ein grosser Tumor vorliegt (≥ 200 ml), welcher wegen primärer oder fehlender Resektion respektive präoperativer Radiotherapie bezüglich des pathologischen Ansprechens nicht evaluiert werden kann. Zudem bei Vorliegen eines unresezierten Tumors < 200 ml mit schlechtem radiologischen Ansprechen auf die Induktionschemotherapie. In dieser Patientengruppe konnte durch die Busulfan und Melphalan Hochdosischemotherapie ein klar verbessertes Gesamtüberleben erreicht werden: OS nach 8 Jahren 64.5% (95% CI, 54.4% – 73.5%) versus 55.6% (95% CI, 45.8% – 65.1%) (10).
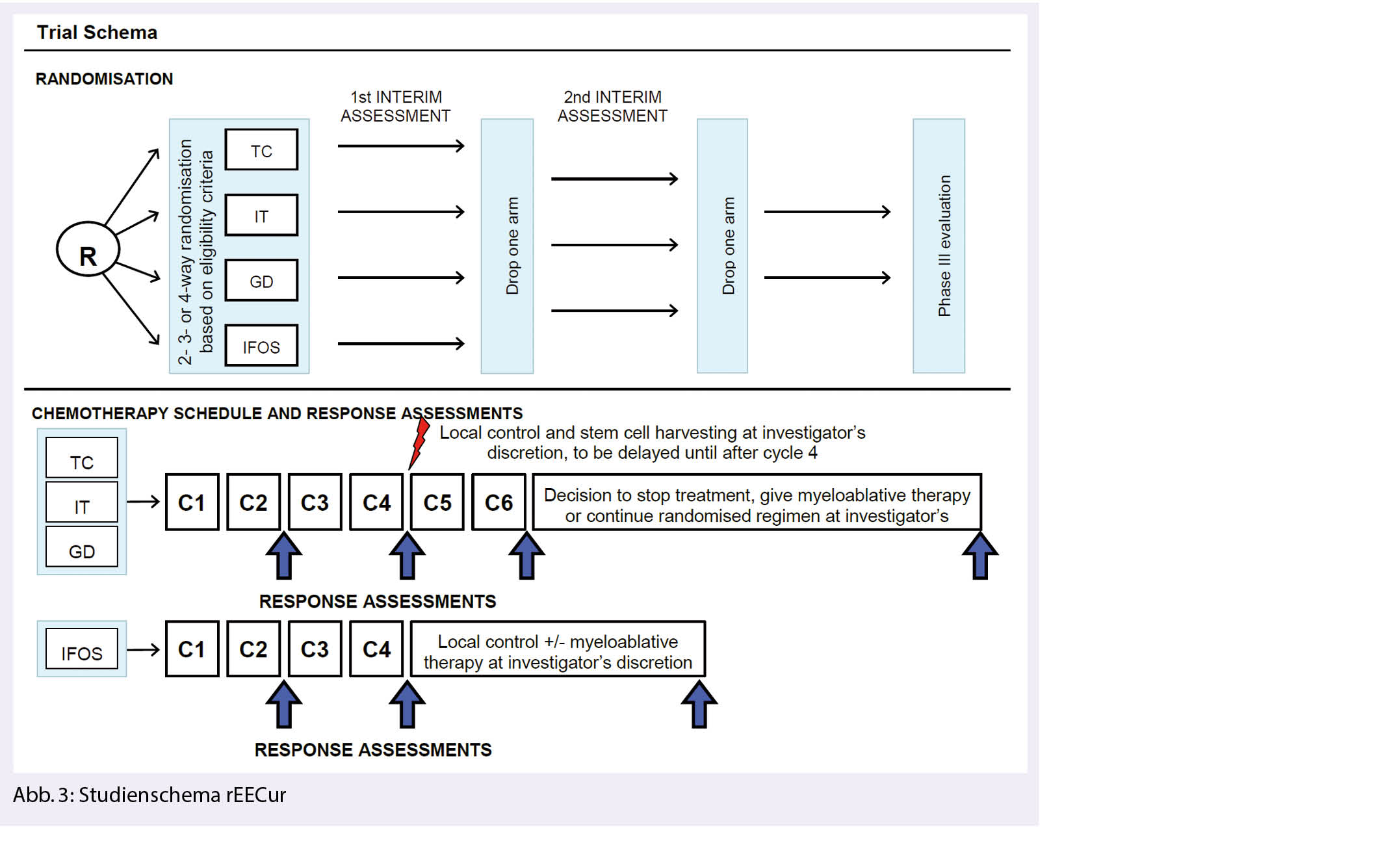
rEECur, Therapie des rezidivierten und primär refraktären Ewing Sarkoms
In der rEECur Studie werden erstmals verschiedene Chemotherapieschemata in der rezidivierten und primär refraktären Situation beim Ewing Sarkom untersucht. Dabei erfolgte initial eine Randomisierung in folgende 4 Arme: Topotecan / Cyclophosphamid (TC), Irinotecan/Temozolomid (IT), Gemcitabin/Docetaxel (GD) und hochdosiertes Ifosfamid (IFOS) (Abb. 3). In der ersten geplanten Interimsanalyse zeigte sich eine geringere Effizienz der Therapie mit Gemcitabine/Docetaxel verglichen mit den übrigen 3 Armen (RR 11.5%, median PFS 3.0 Monate, medianes OS 13.7 Monate), sodass dieser Arm geschlossen wurde (11). In der zweiten Interimsanalyse war nun der IT-Arm unterlegen mit einer Ansprechrate von 20%, einem medianen progressionsfreien Überleben (progression free survial, PFS) von 4.7 Monaten und einem medianen Gesamtüberleben von 13.9 Monaten (12). In die beiden verbleibenden Arme (TC und IFOS) können weiterhin Patienten eingeschlossen werden.
In der Schweiz wird die Studie durch die Schweizerische Pädiatrische Onkologie Gruppe (SPOG) durchgeführt, ein Einschluss von Patienten > 18 Jahren in SPOG-verantworteten Studien ist bislang nicht möglich. Für seltene Erkrankung wie das Ewing Sarkom ist zwingend, möglichst allen Patienten den Zugang zu Studien zu ermöglichen. Für altersübergreifende Erkrankungen sollte hierfür rasch eine möglichst administrationsarme Lösung zwischen Schweizerische Arbeitsgemeinschaft für Klinische Krebsforschung (SAKK) und der SPOG gefunden werden.
Dies erhält zusätzlich Gewicht, gerät die Altersgruppe der Adoleszenten und jungen Erwachsenen (Adolescents and Young Adults, AYA, Alter 15 – 39 Jahre) zunehmend in den Fokus. Die dieser Altersgruppe eigenen oft komplexen medizinischen, psychologischen und sozialen Aspekte bedürfen sowohl in Studien wie im klinischen Alltag einer gesonderten Betrachtung. So ist es auch eine Tatsache, dass die Studienteilnahme von Patienten dieses Alters oft geringer ist als bei pädiatrischen Patienten resp. älteren Erwachsenen. Dies nicht zuletzt auf Grund der herkömmlich limitierten Zusammenarbeit zwischen Studiengruppen der pädiatrischen und der Erwachsenen Onkologie (13, 14).
Perioperative Systemtherapie
Die Resultate einer japanischen Phase II/III Studie zur perioperativen Systemtherapie beim Weichteilsarkom stützen erneut die Wirksamkeit einer entsprechenden Behandlung (15). In dieser randomisierten Non-Inferioritätstudie wurde bei operablen G2/G3 Weichteilsarkomen eine perioperative Therapie mit Doxorubicin/Ifosfamid 10g/m2 (AI) mit Gemcitabine/Docetaxel (GD) verglichen. Es erfolgte die Verabreichung von 3 Zyklen präoperativ und 2 Zyklen postoperativ. In der zweiten geplanten Interimsanalyse zeigte sich ein 2-Jahresüberleben für AI von 94.3%, für GD von 91.6%. Das PFS für AI lag bei 81.9%, für GD bei 64% bei einer HR von 2.32 (95% CI 1.22 – 4.39). Die Studie wurde auf Grund der fehlenden Non-Inferiorität von GD geschlossen. Somit bleibt AI Standard für eine perioperative Systemtherapie, falls eine solche durchgeführt wird (16). Die Resultate sind erneut kein formaler Beweis für die Wirksamkeit von AI (neo)adjuvant, legen eine mindestens PFS-relevante Wirkung jedoch sehr nahe – es sei denn, eine Therapie mit GD stellt einen PFS-relevanten Nachteil dar.
Sehr spannend in diesem Kontext der perioperativen Behandlung sind auch die präliminären Daten einer Phase II Studie zur konkomitierenden neoadjuvanten Checkpoint Blockade (Nivolumab oder Ipilimumab / Nivolumab) mit Radiotherapie bei dedifferenziertem retroperitonealen Liposarkom (DDLPS) und undifferenziertem pleomorphen Sarkom (UPS) der Extremiät oder des Rumpfs (17). Es zeigte sich beim UPS ein sehr hohes pathologisches Ansprechen von 95% (95% CI 85% – 99%), beim DDLPS von 22.5%. Dies unterstreicht die Wertigkeit der Checkpointinhibitortherapie bei gewissen Sarkomentitäten. Weitere zu beachten ist, dass das pathologische und radiologische Ansprechen gemäss RECIST Kriterien nicht korrelieren – diese erweisen sich somit zur Evaluation des Therapieansprechens bei Sarkomen erneut als ungenügend.
Ebenso erfreulich sind die Resultate der TRASTS Studie, in welcher Patienten mit myxoidem Liposarkom eine präoperative Radiochemotherapie erhielten: 3 Zyklen Trabectedin sowie Bestrahlung mit 45 Gy (18). Diese Kombination liegt bei bekannter Sensitivität des myxoiden Liposarkoms sowohl auf Radiotherapie als auch auf Trabectedin sowie radiosensitivierender Wirkung von Trabectedin nahe. Im Resektat zeigte sich bei 23/43 Patienten residueller Tumor >10%, in den restlichen 47% ≤ 10% respektive bei 14% der Patienten (6/43) ein komplettes pathologisches Ansprechen.
Fortgeschrittenes Sarkom
Für die Behandlung fortgeschrittener Sarkome wurden am ASCO 2020 hauptsächlich Daten zu Kombinationstherapien bereits bekannter Substanzen präsentiert. Weiterhin eher im Hintergrund steht die Immuntherapie, wenn diese auch in einzelnen Entitäten gute Ansprechraten aufweist, wie bereits erwähnt.
So bestätigt die finale Analyse der LMS-02 Studie mit den OS-Daten die Wirksamkeit einer Kombinationstherapie mit Doxorubicin und Trabectedin beim Leiomyosarkom (LMS). Diese Phase II Studie prüfte beim uterinen LMS (u-LMS) wie beim Weichgewebs LMS (st-LMS) eine Therapie mit Doxorubicin + Trabectedin als Erstlinientherapie. Es zeigte sich ein Ansprechen (complete remission, CR und partial remisson, PR) für das u-LMS von 59.6% (47 Patienten), für das st-LMS von 39.4% (61 Patienten) (19). Nach einem medianen Follow-up von 7.2 Jahren betrug das mediane PFS 10.1 Monate und das mediane OS 34.4 Monate (20). Im indirekten Vergleich fand sich in der Phase III Announce Studie im Therapiearm Doxorubicin mono beim metastasierten LMS ein PFS von 6.9 Monaten und ein OS von 21.9 Monaten. Eine entsprechende randomisierte Phase III Studie läuft (LMS04).
Eine Phase Ib/II Studie zur Kombinationstherapie mit Lenvatinib und Eribulin beim fortgeschrittenen Lipo- und Leiomyosarkom nach maximal zwei Vortherapielinien weckt weitere Hoffnung, erreichte die Ansprechrate gemäss CHOI Kriterien doch sehr gute 67% (Eribulin mono overall response rate 4%) mit einem medianen progressionsfreien Überleben von 56 Monaten (Eribulin mono 2.6 Monate) (21).
Von der FDA wurden im letzten Jahr zudem verschiedene neue Substanzen für die Behandlung von Sarkomen zugelassen:
2019 Entrectinib, nach Larotrectinib der zweite TRK-Inhibitor für NTRK-fusionsassoziierte Tumore, basierend auf den drei zulassungsrelevanten Studien ALKA, STARTRK-1 und STARTRK-2 sowie Pexidartinib (CSF-1 Rezeptor Inhibitor) für die Behandlung tenosynovialer Riesenzelltumore (22).
2020 erfolgte bislang die Zulassung von Tazemetostat (EZH Inhibitor) für epithelioide Sarkome (23) sowie von Pomalidomid für das HIV-positive wie -negative Kaposi Sarkom (accelerated approval, basierend auf der NIH Studie 12-C-0047). Weiter zugelassen wurde Selumetinib, ein MEK-Inhibitor, für die Behandlung plexifomer Neurofibrome der Neurofibromatose Typ 1 (24), sowie in der Therapie des gastrointestinalen Stromatumors (GIST) Avapritinib (25), mit Wirksamkeit bei PDGFRA D842V Mutation, sowie Ripretinib (26) in der späteren Therapielinie.
Die Behandlung der Sarkome als Gesamtentität gehört der Vergangenheit an. Digitalisierung mit verstärkter elektronischer Vernetzung sowie die Behandlung der Patienten in national und international kollaborierenden Netzwerken bilden die Voraussetzung, den therapeutischen Fortschritt dieser teils ultrararen Erkrankungen möglichst rasch weiter voranzutreiben.
Copyright bei Aerzteverlag medinfo AG
Klinik für Medizinische Onkologie und Hämatologie
Kantonsspital St.Gallen
Rorschacher Strasse 95
9007 St.Gallen
Leitender Arzt / Leitung Ambulatorium St. Gallen & Rorschach
Kantonsspital St. Gallen
Klinik für Med. Onkologie/Hämatologie
Rorschacher Strasse 95
9007 St. Gallen
Die Autoren haben im Zusammenhang mit diesem Artikel keine Interessenskonflikte deklariert.
1. Kollar A et al. Incidence, mortality, and survival trends of soft tissue and bone sarcoma in Switzerland between 1996 and 2015. Cancer Epidemiol. 2019; 63: 101596.
2. Loong et al. International collaborations and regional challenges in providing specialist multidisziplinary sarcoma care. ASCO Educational Book 2019
3. Blay J-Y et al. Surgery in Reference Centers Improves Survival of Sarcoma Patients: A Nationwide Study. Ann Oncol 2019; 30: 1143.
4. Palmerini E et al. Graceful Project: a global collaboration on CIC-DUX4, BCOR-CCNB3, high grade undif-ferentiated round cell sarcoma (URCS), CTOS 2019
5. Johansson B et al. Most gene fusions in cancer are stochastic events. Genes Chromosomes Cancer 2019; 58: 607.
6. Brennan B et al. Comparison of chemotherapy regimens in Ewing sarcoma (ES): Overall and subgroup results of the Euro Ewing 2012 randomized trial (EE2012). J Clin Oncol 2020; 38 (suppl; abstr 11500).
7. Anderton J et al. International randomised controlled trial for the treatment of newly diagnosed EWING sarcoma family of tumours. Trials 2020; 21: 96.
8. Gorlick R et al. Dose Intensification Improves the Outcome of Ewing Sarcoma.
J Clin Oncol 2018; 36: 3072.
9. Dirksen U et al. Efficacy of add-on treosulfan and melphalan high-dose therapy in patients with high-risk metastatic Ewing sarcoma: Report from the International Ewing 2008R3 trial. J Clin Oncol 2020; 38 (suppl; abstr 11501).
10. Wehlan J et al. High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J Clin Oncol 2018; 36: 3110.
11. McGabe M et al. Results of the first interim assessment of rEECur, an international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma.J Clin Oncol 2019; 37 (suppl; abstr 11007).
12. McGabe M et al. Results of the second interim assessment of rEECur, an international randomized con-trolled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma (RR-ES). J Clin Oncol 2020; 38 (suppl; abstr 11502).
13. Osborn M et al. Models of care for adolescent and young adult cancer programs. Pediatr Blood Cancer 2019; 66: e27991.
14. Reed DR et al. Sarcoma as a Model for Adolescent and Young Adult Care. J Oncol Pract 2019; 15: 239.
15. Tanaka K et al. Results of a randomized phase II/III study comparing perioperative adriamycin plus ifosfamide and gemcitabine plus docetaxel for high-grade soft tissue sarcomas: Japan Clinical Oncology Group study JCOG1306. J Clin Oncol 2020; 38 (suppl; abstr 11504).
16. Rothermundt C. Perioperative treatment of soft-tissue sarcoma. Memo 2020;13:174.
17. Roland CL et al. Preliminary results of a phase II study of neoadjuvant checkpoint blockade for surgically resectable undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated liposarcoma (DDLPS). J Clin Oncol 2020; 38 (suppl; abstr 11505.
18. Gronchi A et al. Trabectedin and radiotherapy in soft-tissue sarcoma (TRASTS) study: An international, prospective, phase II trial in localized myxoid liposarcoma—A collaborative Spanish (GEIS), Italian (ISG) and French (FSG) group study. J Clin Oncol 2020; 38 (suppl; abstr 11514).
19. Pautier P et al. Trabectedin in combination with doxorubicin for first-line treatment of advanced uterine or soft-tissue leiomyosarcoma (LMS-02): a non-randomised, multicentre, phase 2 trial. Lancet Oncol 2015; 16: 457.
20. Patricia Pautier A single-arm multicenter phase II trial of doxorubicin (Doxo) in combination with trabecte-din (Trab) given as first-line treatment to patients with metastatic/advanced uterine (U-LMS) and soft tissue leiomyosarcoma (ST-LMS): Final results of the LMS-02 study J Clin Oncol 2020; 38 (suppl; abstr 11506).
21. Tom Wei-Wu Chen et al. A Ib/II study of the combination of lenvatinib (L) and eribulin (E) in advanced liposarcoma (LPS) and leiomyosarcoma (LMS) (LEADER).
J Clin Oncol 2020; 38 (suppl; abstr 11507)
22. Tap WD et al. Pexidartinib versus placebo for advanced tenosynovial giant cell
tumour (ENLIVEN): a randomised phase 3 trial. Lancet. 2019; 394: 478.
23. Stacchiotti S et al. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients with epi-thelioid sarcoma. J Clin Oncol 2019; 37 (suppl; abstr 11003).
24. Gross AM et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Engl J Med 2020; 382:1430
25. Heinrich M et al. Clinical activity of avapritinib in ≥ fourth-line (4L+) and PDGFRA Exon 18 gastrointestinal stromal tumors (GIST). J Clin Oncol 2020; 38 (suppl 4; abstr 826)
26. Blay J-Y et al. Ripretinib in Patients With Advanced Gastrointestinal Stromal Tumours (INVICTUS): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol 2020; 21: 923


info@onco-suisse
- Vol. 10
- Ausgabe 6
- Oktober 2020