
Die Sichelzellkrankheit ist eine angeborene Hämoglobinopathie bedingt durch eine Mutation der ß-Kette des Hämoglobins, dem Hämoglobin S. Bei Sauerstoffmangel kommt es zur Sichelung der Erythrozyten, was eine Adhäsion an das Gefässendothel begünstigt und damit zu akuten Komplikationen der Vasookklusion und zu chronischen Organschäden führt. Die klinischen Manifestationen und die Prognose sind variabel. Das klinische Management ist häufig anspruchsvoll.
Sickle cell disease is a congenital hemoglobinopathy caused by a mutation of the ß-chain of hemoglobin, the hemoglobin S. In case of oxygen deficiency sickling of the erythrocytes occurs, which promotes adhesion to the vascular endothelium and thus leads to acute complications of vaso-occlusion and chronic organ damage. The clinical picture and the prognosis are variable. The clinical management is often challenging.
Keywords: Sickle cell disease, hemoglobinopathy, mutation of the ß-chain, sickling of the erythrocytes
Es wird geschätzt, dass ungefähr 7% der Weltbevölkerung eine monogenetische Hämoglobinkrankheit trägt. Dazu gehört die Sichelzellkrankheit (Sickle Cell Disease, SCD). Über 250’000 Kinder mit einer klinisch relevanten SCD Form werden jährlich geboren (1).
Die Prognose der SCD hat sich in den letzten Jahrzehnten wesentlich verbessert. Das mediane Überleben beträgt bis 60-70 Jahre für Patienten, welche in einkommensstarken Ländern leben (2). Deshalb ist die SCD auch bei uns nicht mehr eine hauptsächlich pädiatrische Entität, sondern betrifft viele Erwachsene. Die Klinik der SCD ist sehr variabel, kann aber potentiell tödliche Folgen haben.
Definition und Epidemiologie
Die SCD ist eine autosomal-rezessive Erbkrankheit mit dem Austausch von Glutaminsäure gegen Valin an der Position 6 der ß-Kette des Hämoglobins (Hb). Dadurch entsteht anstelle des adulten HbA das HbS.
In gesunden Individuen, setzen sich die Hb-Ketten aus HbA (α2ß2) zu 96-97%, HbA2 (α2δ2) zu 2-3,5% und HbF (α2γ2) <1% zusammen (Tab. 1). Bei der SCD wird das HbA komplett oder teilweise durch das HbS ersetzt.
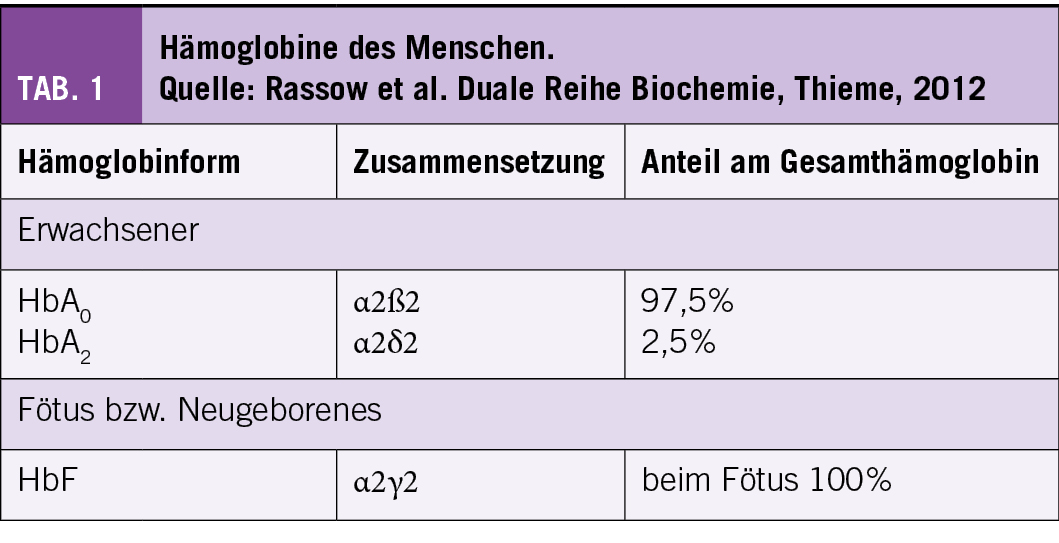
Je nach Erbmuster und Kombination mit anderen Mutationen des ß-Gens kommt es zu unterschiedlichen Genotypen und Phänotypen der SCD. Die klinisch wichtigsten Formen der SCD sind:
• Die homozygote HbSS: Beide ß-Gene sind mutiert und es werden einzig S-Ketten produziert. Dies ist die häufigste Form.
• Die Compound-heterozygote SCD-Syndrome (Kombinationen von HbS-Mutation mit einer anderen ß-Genmutation):
i. Die HbSß° Thalassämie: es wird keine ß-Kette und lediglich eine S-Kette produziert; klinisch ähnlich zum HbSS-Typ.
ii. Die HbSß+ Thalassämie: Produktion von HbS und wenig residuelle Produktion von ß-Ketten.
iii. Die HbSC: Hier liegen S- und C-Ketten vor, keine normalen ß-Ketten.
Die heterozygote Form (HbA/S) hat in der Regel keinen Krankheitswert (3-5) (Abb. 1).
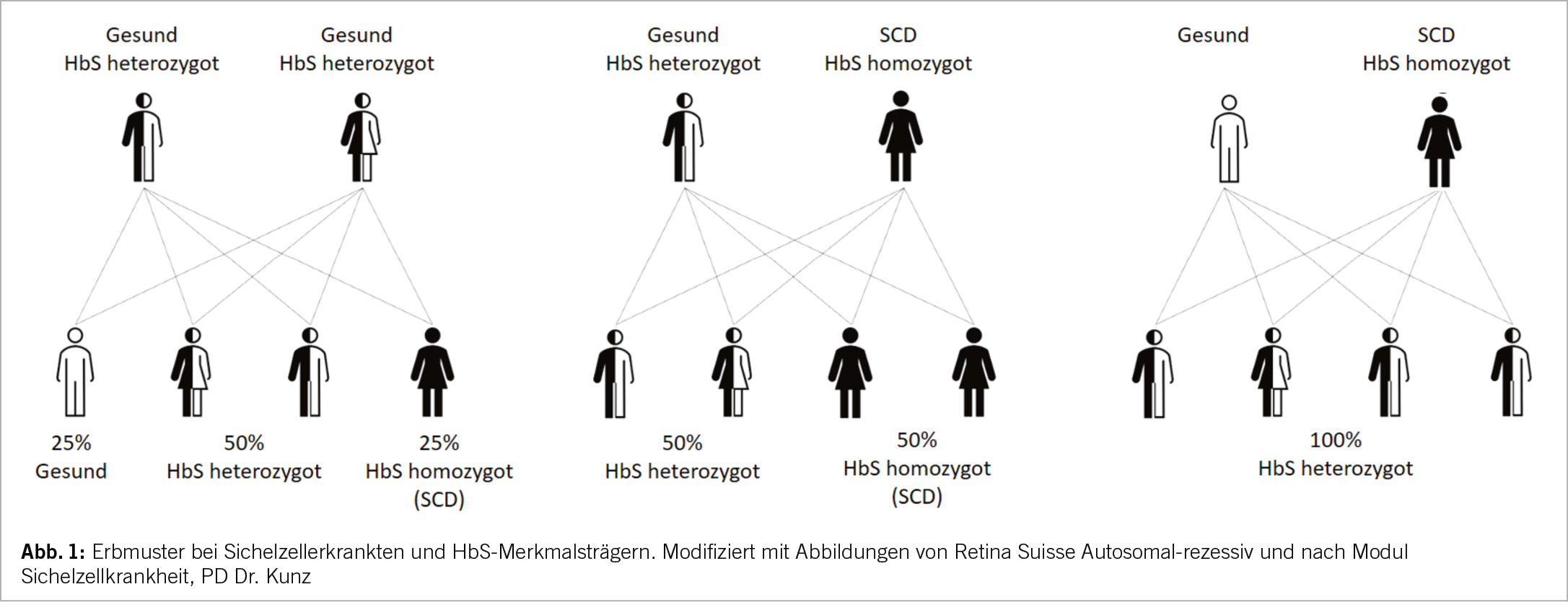
Die SCD ist hauptsächlich in Malariagebieten verbreitet, insbesondere in den tropischen Anteilen Afrikas, aber auch in Indien und in der arabischen Halbinsel (Abb. 2). Vermutlich stellt die SCD mit Ausnahme des HbSS-Phänotyps einen Selektionsvorteil gegenüber schweren Verläufen der Malaria dar (4). Durch Migration leben immer mehr Patienten mit SCD auch in Nord- und Südamerika sowie in Europa. Es wird geschätzt, dass in der Schweiz etwa 400-600 Menschen an SCD leiden. Die Dunkelziffer könnte dennoch höher sein, da es kein systematisches Screening oder Register gibt (6).
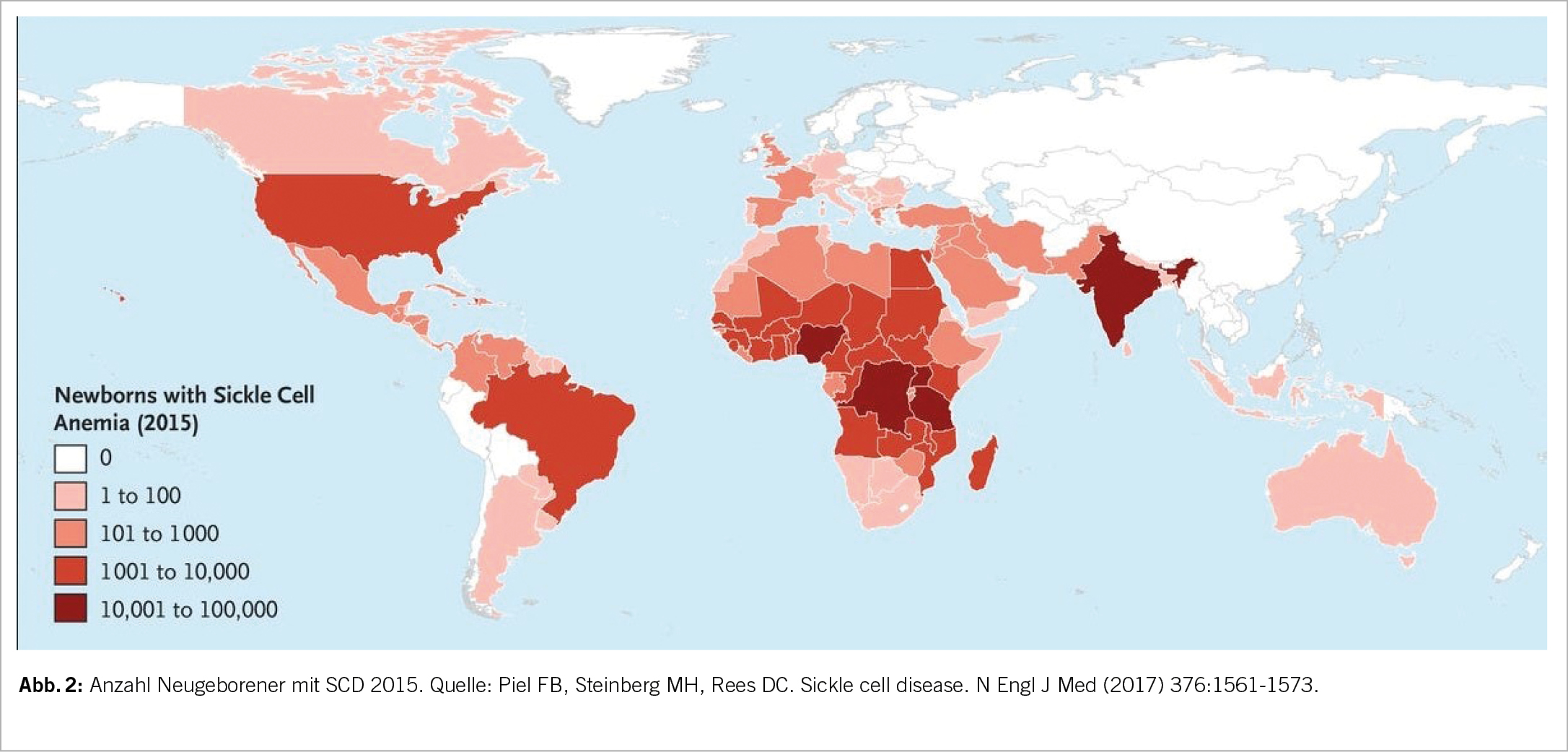
Pathophysiologie
Bei Sauerstoffmangel formt das abnorme HbS Polymere, welche mit der Erythrozytenmembran interagieren und zu einer sichelzellförmigen Deformation des Erythrozyten führen (Abb. 3). Durch die Deformierung («Sickling») entsteht eine Vasookklusion insbesondere in der Mikrozirkulation. Mit rezidivierenden vasookklusiven Krisen kommt es zu einem Endothelschaden der Gefässe und zu wiederholten lokalen Ischämien. Die Lebenszeit der Erythrozyten nimmt aufgrund des mechanischen Schadens ab, eine chronische Hämolyse ist die Folge. Durch die Hämolyse und das konsekutiv freie Hb entstehen Sauerstoffradikale. Die Kaskade führt zu einem prothrombotischen und inflammatorischen Zustand. Die Ischämie und Hämolyse führen zu einem Multiorganschaden (5, 7).
Diagnose
Zentral ist die Anamnese (Familienanamnese, Symptome, Anzahl vasookklusiver Krisen, Trigger, Transfusionen, Thromboembolien) sowie klinischer Status mit Fokus auf die Neurologie und Suche einer Splenomegalie.
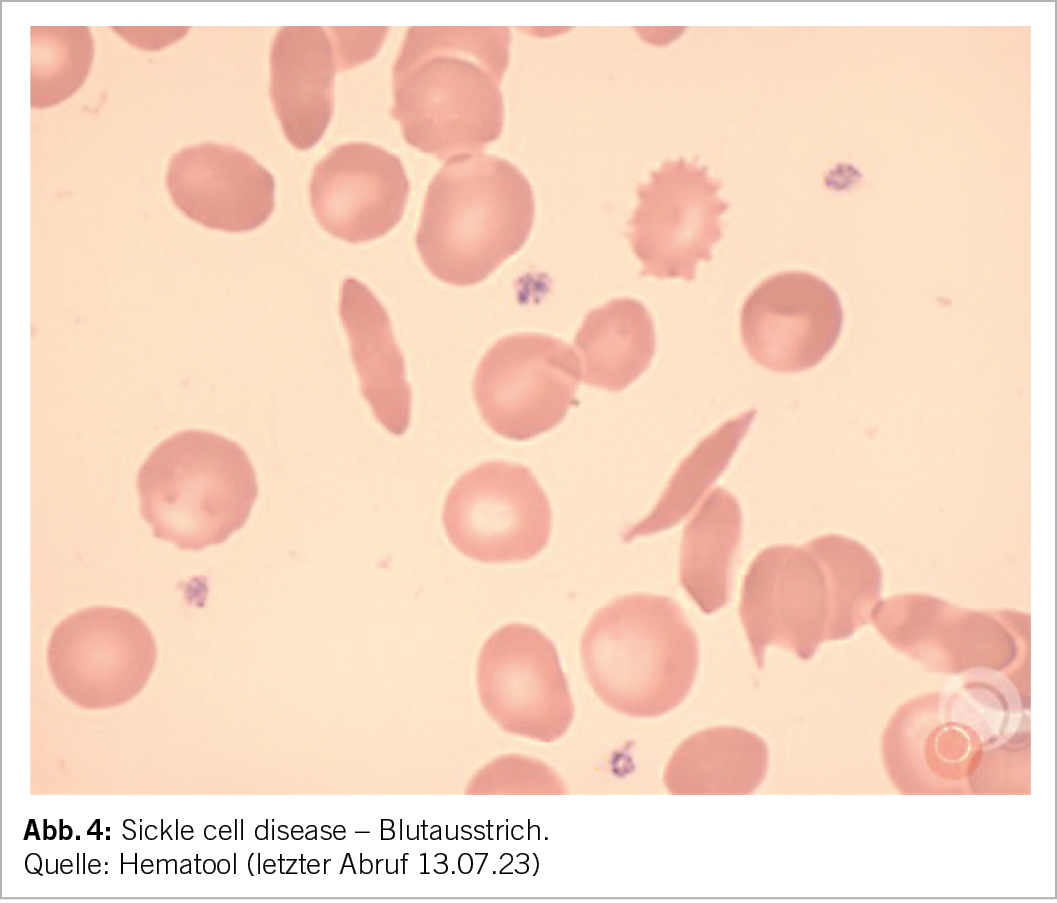
Im Blutbild zeigt sich meist eine mittelschwere (Hb 85-100 g/l) normozytäre oder makrozytäre (aufgrund der Retikulozytose) regenerative Anämie, wobei eine Mikrozytose (bei Eisenmangel oder bei kombinierter heterozygoten alpha-Thalassämie) auch möglich ist. Die Anämie ist je nach Phänotyp unterschiedlich ausgeprägt. Im Blutausstrich zeigen sich Sichelzellen, Howell-Jolly Körperchen (Funktionseinschränkung der Milz), Erythroblasten, sowie bei kombinierter Thalassämie zusätzlich Targetzellen (Abb. 4).
Im Chemogramm finden sich Hämolyseparameter (erhöhte LDH, erhöhtes Bilirubin, vermindertes Haptoglobin).
Mittels Chromatographie (HPLC) oder Hb-Elektrophorese führt die Messung des HbS, HbA, HbA2, HbF und HbC zur definitiven Diagnose.
Weiter wird ein abdominaler Ultraschall zur Beurteilung der Milz, Leber, Gallenblase und Nieren empfohlen sowie ein TTE zur Evaluation einer pulmonalen Hypertension und der kardialen Funktion. Eine Bildgebung des Kopfes zur Beurteilung von stattgehabten, häufig asymptomatischen Schlaganfällen (silent strokes) und eine augenärztliche Untersuchung (proliferative Retinopathie) sollte durchgeführt werden (5).
Klinisches Bild
Die Beschwerden sind je nach Phänotyp unterschiedlich schwer ausgeprägt. In Individuen mit der homozygoten HbSS Krankheit und HbS/ß°-Form, treten erste Beschwerden meist einige Monate nach der Geburt auf, nach dem Wechsel von HbF auf HbA/HbS.
Aufgrund der komplexen Pathophysiologie kommt es zu einer Vielzahl von akuten und chronischen Komplikationen (Abb. 5). Zu den klassischen klinischen Erscheinungen gehören die vasookklusiven Schmerzkrisen, häufig mit Osteonekrosen assoziiert, die chronische hämolytische Anämie, Infektionen aufgrund der funktionellen Asplenie und das akute Thoraxsyndrom. Weitere chronische Organschäden können sich asymptomatisch im Laufe der Jahre entwickeln (z.B. Nephropathie, Herzinsuffizienz, pulmonal arterielle Hypertension).
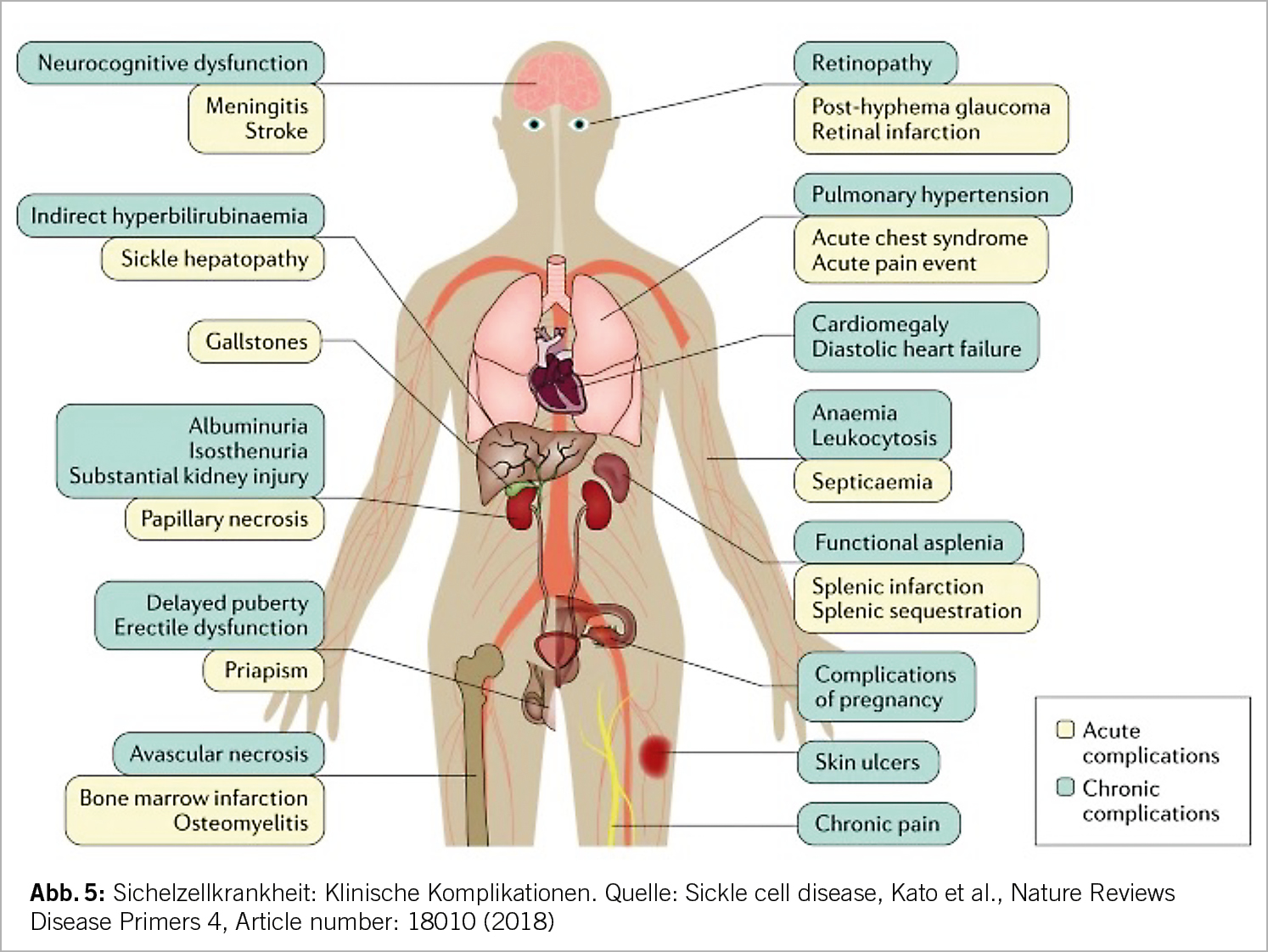
Vasookklusive Krisen werden getriggert durch Infektionen, Azidose, Dehydratation, Hypothermie und Hypoxie. Die damit einhergehende lokale Ischämie führt zu akuten und starken Schmerzen. Diese Durchblutungsstörungen können alle Organe betreffen und zu einer entsprechenden Problematik führen: beim Gehirn kommt es zu stummen oder symptomatischen Infarkten, Knochennekrosen entstehen, die Folge der Lungenbeteiligung ist eine pulmonale Hypertonie bis hin zum respiratorischen Versagen, in den Nieren führen Vasookklusionen zu einer Glomerulopathie, im Auge bildet sich eine proliferative Retinopathie (insbesondere bei HbSC). Rezidivierende Milzinfarkte führen zu einem Funktionsverlust der Milz (funktionelle Asplenie), welche bei vielen Patienten bereits seit der Kindheit besteht. Dies geht einher mit einer erhöhten Infektanfälligkeit vor allem durch Pneumokokken, Hämophilus, Meningokokken.
Aufgrund der multiplen chronischen Organveränderungen ist die SCD eine Multisystemerkrankung mit einhergehender erhöhter Morbidität und Mortalität sowie reduzierter Lebensqualität (5, 7, 8).
Therapie
Allgemeine Massnahmen
Die Betreuung von Patienten mit SCD erfordert die regelmässige Evaluation der physischen und psychischen Gesundheit sowie der Lebensqualität und der sozialen Integration. Dadurch ist die Behandlung dieser Patienten häufig anspruchsvoll und erfolgt idealerweise multidisziplinär. Ebenfalls von zentraler Bedeutung ist der Übertritt der Patienten von der Pädiatrie zur Erwachsenenmedizin (Transition).
Zum allgemeinen Management gehört eine gute Schulung der Patienten um Trigger zu vermeiden (z.B. physische Überanstrengung, Dehydrierung, Kälteexposition). Leichte Schmerzen werden meist selbst behandelt. Bei Fieber, Infekten oder starken Schmerzen muss eine umgehende Vorstellung in einem Zentrumspital erfolgen (4, 5, 7, 8).
Medikamentöse Therapie
Die therapeutischen Ansätze für die SCD sind eine Stimulation der Produktion des HbF, eine Verbesserung der Durchblutung in den kleinen und mittleren Gefässen und eine Erhöhung der Sauerstoffbindung an das Hb.
Hydroxycarbamid/Hydroxyurea (Lialir®, Siklos®) ist ein altes Zytostatikum, welches die Produktion von HbF steigert und damit die Polymerisation von HbS reduziert. Der Einsatz von Hydroxyurea bereits ab dem ersten Lebensjahr gehört zur Standardtherapie. Es ist das einzige in der Schweiz zugelassene Medikament für die SCD. Bei Patienten unter Hydroxyurea sollte alle 3 – 4 Monate der Anteil an HbF gemessen werden (idealerweise > 20%) (4, 5, 7, 8).
Crizanlizumab ist ein monoklonaler Antikörper gegen den P-Selektin-Rezeptor, welcher sowohl auf Blutzellen als auch auf Endothelien exprimiert ist. Durch die Bindung von Crizanlizumab an die Endotheloberfläche wird die Adhäsion zwischen Blutzellen und Endothel reduziert und somit auch lokale Ischämien und vasookklusive Krisen. Crizanlizumab (Adakveo®) ist aktuell nur direkt vom Hersteller (Novartis) und im Rahmen eines Managed Access Program erhältlich (9).
Voxelotor ist ein Hb-Modulator, welcher die Hb-Sauerstoffaffinität erhöht und die Polymerisation reduziert. Das Medikament ist in der Schweiz nicht zugelassen (10).
Behandlung der akuten Symptome
Bei akuten schweren Schmerzkrisen benötigt es eine intravenöse Hydrierung, Sauerstoffgabe, eine ausreichende Analgesie (meist Opiatpräparate), die Behandlung von Trigger, wenn bekannt, sowie rascher Beginn einer antibiotischen Therapie bei Fieber. Aufgrund des erhöhten thromboembolischen Risikos ist an eine Prophylaxe zu denken. Zu den gefährlichsten Komplikationen gehört das akute Thoraxsyndrom (acute chest syndrome), häufig vergesellschaftet mit einer Lungenembolie und/oder bakterieller Pneumonie. Dies ist die häufigste Todesursache. Eine akute Milzsequestration (Pooling von Blut in der Milz) mit Hb-Abfall bis zum hypovolämen Schock ist selten, erfordert aber auch eine intensivmedizinische Behandlung.
Bei chronischer Hämolyse und damit einhergehend erhöhtem Bedarf an Folsäure wird eine entsprechende Dauersubstitution empfohlen.
Aufgrund der funktionellen Asplenie ist es wichtig, dass Erkrankte sich gegen Pneumokokken, Meningokokken, Hepatitis B, SARS-CoV-2 und saisonale Influenza impfen. Ein Asplenie-Ausweis muss ausgehändigt werden (4, 5, 7, 8).
Transfusionen
Einfache Transfusionen oder Austauschtransfusionen (z.B. 2x Phlebotomie von 400-500 ml alternierend mit Transfusion von 2 Erythrozytenkonzentraten) verbessern die Sauerstoffkapazität und führen zu einer Verminderung des HbS. In speziellen Situationen, wie z.B. als Vorbereitung für einen grossen chirurgischen Eingriff oder einen Kaiserschnitt, und in spezialisierten Zentren ist ein maschineller Austausch mittels Erythrozenapherese indiziert.
Es sollte ein Hb zwischen 90-100 g/l angestrebt werden. Chronisch tiefere Hb Werte < 80 g/l können lokale Ischämien begünstigen, insbesondere im Gehirn, anderseits Hb Werte > 110 g/l können durch Hyperviskosität die Mikrozirkulation beeinträchtigen. Ein HbS ≤ 30% ist das Ziel, sowohl für die Behandlung im chronischen Setting als auch bei akuten Krisen, wobei dieser Wert mit manuellen Austauschtransfusionen nur schwierig zu erreichen ist (4, 5, 7, 8).
Allogene Stammzelltransplantation
Die Stammzelltransplantation mit HLA-identischen Geschwisterspendern zeigt im Kindesalter gute Resultate. Leider sind aber Geschwister häufig auch von der SCD befallen und kommen als Spender nicht in Frage. Passende Fremdspender sind ebenfalls selten im Spenderregister zu finden, da die meisten Patienten afrikanischer Abstammung und nur wenige Gesunde ähnlicher Herkunft als freiwillige Stammzellspender registriert sind (3, 4, 6, 8).
Zukunft Gentherapien
In verschiedenen zurzeit laufenden klinischen Studien wird der Einsatz von autologen Stammzellen untersucht, bei welchen mittels Gentherapien die mit HbS assoziierte Genmutation korrigiert wird (11).
Copyright bei Aerzteverlag medinfo AG
Klinik für Hämatologie, Universitätsspital Basel
– Klinik für Hämatologie, Universitätsspital Basel
– Blutspendezentrum SRK beider Basel
Die Autorinnen haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Hemoglobinopathies: clinical manifestations, diagnosis, and treatment; Kohne Elisabeth, Dtsch Arztebl Int. 2011 Aug;108(31-32):532-40. doi: 10.3238/arztebl.2011.0532. Epub 2011 Aug 8
2. Sickle Cell Disease, Piel et al. N Engl J Med 2017; 376 :1561-1573
3. https://www.cdc.gov/ncbddd/sicklecell/facts.html (letzter Abruf 13.07.23)
4. Sickle-cell disease, Rees et al. The Lancet, Volume 376, Issue 9757, 11-17 December 2010, Pages 2018-2031
5. Hematool, https://hematool.ch/ (letzter Abruf 13.07.23)
6. https://www.novartis.com/ch-de/news/als-wuerden-die-knochen-von-innen-nach-aussen-explodieren, 15.06.2020
7. Sickle cell disease, Kato et al., Nature Reviews Disease Primers 4, Article number: 18010 (2018)
8. Onkopedia-Leitlinie Sichelzellkrankheiten, https://www.onkopedia.com/de/onkopedia/guidelines/sichelzellkrankheiten/@@guideline/html/index.html (letzter Abruf 13.07.23)
9. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease, Kenneth / Ataga. N Engl J Med. 2017 Feb 2;376(5):429-439. Doi: 10.1056/NEJMoa16117700. Epub 2016 Dec 3.
10. An expert review of voxelotor for the treatment of hemolytic anemia in patients with sickle cell disease: ‘bridging the gap between laboratory data and patient related outcomes’. Baba P D Inusa; Expert Rev Hematol. 2023 Jun22;1-7. Doi: 10.1080/17474086.2023.2226858.
11. Gentherapie von Hämoglobinkrankheiten – aktuelle Konzepte und Herausforderungen, Andreas E. Kulozik, Joachim Kunz. https://www.klinikum.uni-heidelberg.de/fileadmin/kinderklinik/kinderklinik_3/PDF/5_6.Kulozik_Kunz_publication_Ha__moglobinkrankheiten.pdf (letzter Abruf 13.07.23)


info@onco-suisse
- Vol. 13
- Ausgabe 6
- September 2023