
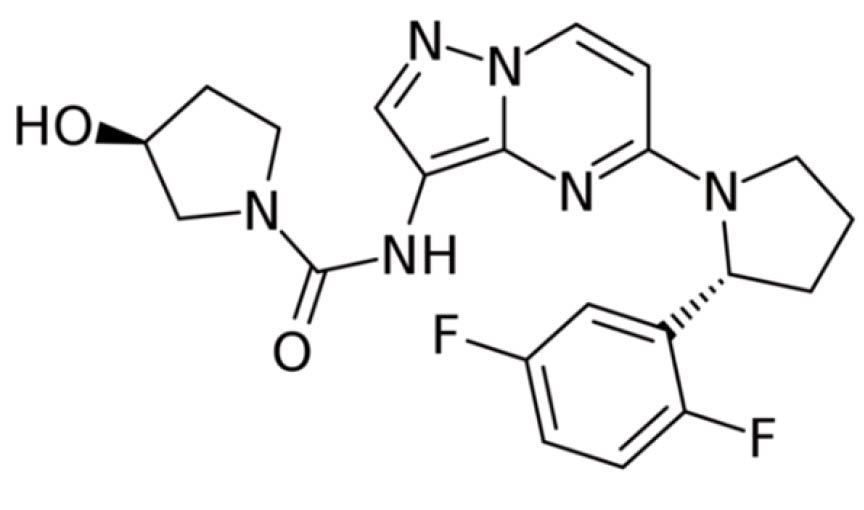
Wirkmechanismus
Larotrectinib ist ein niedermolekularer hochselektiver Inhibitor der drei Tropomyosin-Rezeptor-Kinase-Proteine TRKA, TRKB und TRKC, die zur Schmerzregulierung, Propriozeption, Appetitsteuerung sowie Gedächtnisregulierung beitragen. Bei etwa einem Prozent aller soliden Krebsarten und in gut 1000 Fällen jährlich in Europa liegt ein Fusionsprotein vor, an dem eines der drei Gene der Tropomyosin-Rezeptor-Kinase-Proteine beteiligt ist. Die Gene heissen Neurotrophic receptor tyrosine kinase 1-3, abgekürzt NTRK 1-3. Die TRK-Fusion führt zur Überexpression dieser funktional pathogenen Proteine, was dann dauerhaft Liganden unabhängig den entsprechenden Signalweg aktiviert und ein unkontrolliertes Zellwachstum bewirkt. Daher wird auch von TRK-Fusionstumoren gesprochen.
Für Larotrectinib wurden konzentrationsabhängig eine starke sehr selektive Hemmung der TRK Proteine sowie eine Hemmung der Proliferation von Tumorzellen nachgewiesen. In Xenograft-Mausmodellen mit TRK-Fusion induzierte Larotrectinib eine signifikante Reduktion des Tumorwachstums. Nach Progression unter TRK-Inhibitoren wurden erworbene Resistenzmutationen beobachtet. Larotrectinib hatte minimale Aktivität in Zelllinien mit Punktmutationen in der TRKA-Kinasedomäne, einschliesslich der klinisch nachgewiesenen erworbenen Resistenzmutation G595R. Punktmutationen in der TRKC-Kinasedomäne, die eine klinisch relevante erworbene Resistenz gegenüber Larotrectinib verleihen, sind G623R, G696A und F617L.
Bisher sind ca. dreissig solide Tumorarten und Sarkome bekannt, bei denen TRK-Fusionen wiederholt nachgewiesen wurden. Die meisten gehören zu den Weichteilsarkomen aber auch papillären Schilddrüsenkarzinomen, Speicheldrüsenkarzinomen und Gliomen, für die es international auch bereits Zulassungen gab.
Klinische Wirksamkeit
Da das NTRK-Fusionsprotein bei verschiedenen weiteren Tumoren auftreten kann, wurden 3 Tumortyp-unabhängige sog. Basket-Studies durchgeführt, in die alle Patienten aller Alterskategorien mit einem TRK-Fusionsprotein-positiven Tumor, lokal fortgeschritten oder metastasiert, nach primär erfolgloser Standardtherapie eingeschlossen wurden.
Phase I Studie «LOXO-TRK-14001» (NCT02122913): Offene Dosiseskalations- und expansionsstudie, welche erwachsene Patienten (≥18 Jahre) mit fortgeschrittenen soliden Tumoren einschloss (für die Expansionsphase war das Vorliegen einer NTRK Genfusion erforderlich). Es wurden 8 Patienten in die Wirksamkeitspopulation aufgenommen.
Phase II Studie «NAVIGATE» (NCT02576431): Offene Basket-Studie, welche erwachsene und pädiatrische Patienten (≥12 Jahre) mit fortgeschrittenen soliden Tumoren mit NTRK-Genfusion einschloss. Die verabreichte Dosis betrug 100 mg zweimal täglich. Aus dieser Studie flossen 65 Patienten in die gesamte Wirksamkeitspopulation ein.
Phase I/II Studie «SCOUT» (NCT02637687): Offene Dosiseskalations- und expansionsstudie, welche pädiatrische Patienten (≥28 Tage bis 21 Jahre) mit fortgeschrittenen soliden Tumoren oder primären ZNS-Tumoren einschloss (für die Expansionsphase war das Vorliegen einer NTRK Genfusion erforderlich). Die verabreichte Dosis betrug bis zu 100 mg/m2 zweimal täglich. Aus dieser Studie flossen 38 Patienten in die gesamte Wirksamkeitspopulation ein.
Primärer Endpunkt war die Gesamtansprechrate. In Tabelle 1 werden die Ansprechraten aus den drei Zulassungsstudien in Bezug auf die Art der Isoform des NTRK Fusionsproteins gezeigt.
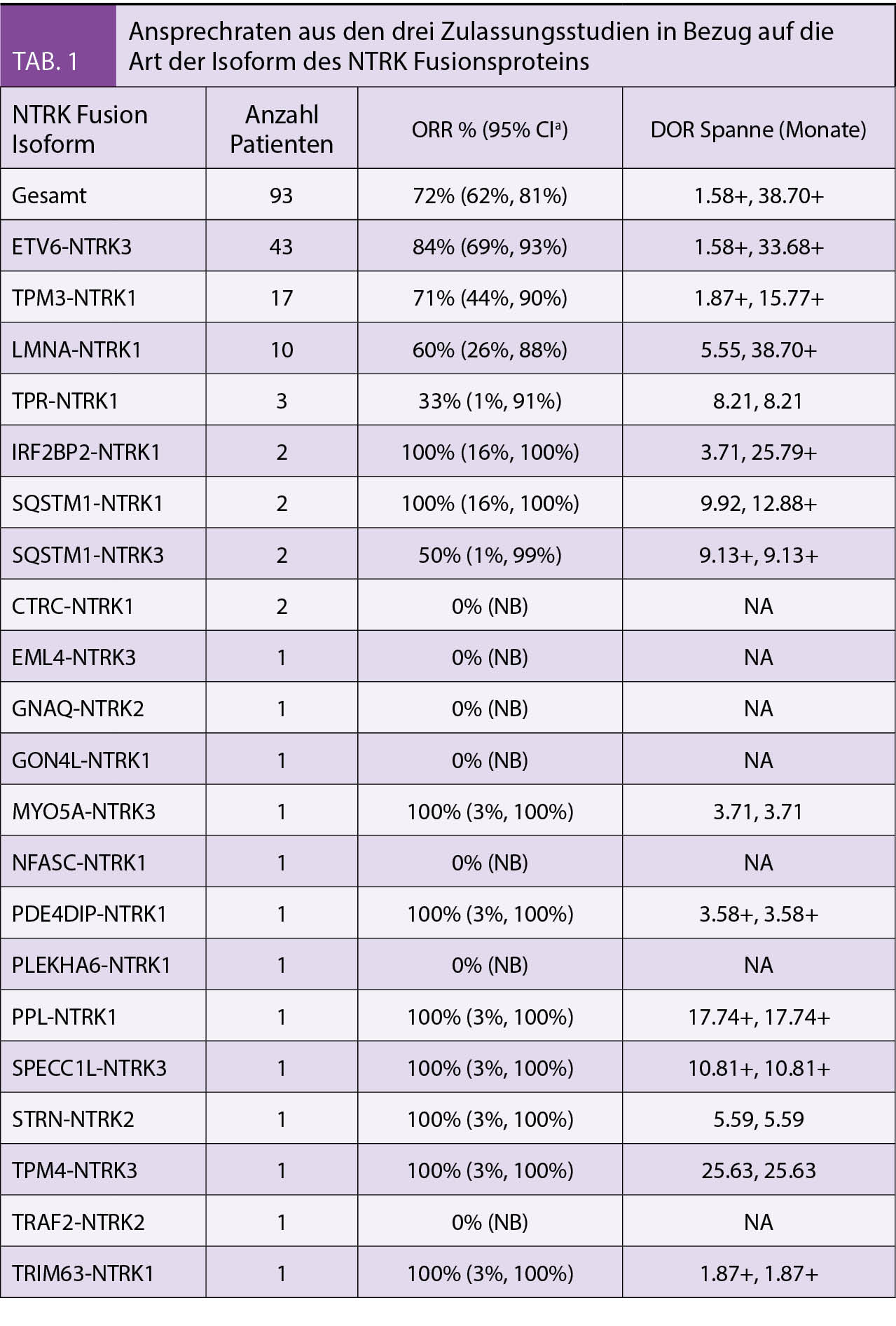
In der Tabelle 2 sind das Ansprechen (ORR) und die Dauer des Ansprechens (DOR) der Patienten aus den drei Zulassungsstudien aufgelistet. Sie zeigten ein hohes und auch lange anhaltendes Ansprechen bei Erwachsenen und Kindern. Die Ansprechrate lag bei 81% für alle Altersgruppen nach medianen 7-18 Monaten und bei Kindern lag das Ansprechen bei 94%. Bei den 34 Kindern wurde in 35% eine komplette Remission, bei 59% eine partielle Remission und 6% eine Stabilisierung beobachtet.
Die Baseline-Charakteristika für die 18 Patienten mit primären ZNS-Tumoren mit einer NTRK-Genfusion, welche nicht in dieser Tabelle angeführt sind, lauten wie folgt: medianes Alter 10 Jahre (1-79 Jahre); 14 Patienten <18 Jahre, 4 Patienten ≥18 Jahre, 13 weiss, 8 männlich und 10 weiblich. Folgende Tumor-Typen lagen vor: Glioblastom (6 Patienten), Gliom (4 Patienten), Glioneural (3 Patienten), nicht spezifiziert (3 Patienten) und Astrozytom (2 Patienten).
Alle Patienten hatten zuvor eine Behandlung für ihre Krebserkrankung erhalten (definiert als chirurgischer Eingriff, Radiotherapie oder systemische Therapie). Im Median hatten die Patienten 1 vorheriges systemisches Therapieregime erhalten.
Baseline-Charakteristika waren:
Folgende NTRK Genfusions-Isoformen sind bei den primären ZNS-Tumoren bestimmt worden: BCR-NTRK2 (bei 3 Patienten), SPECC1L-NTRK2 (bei 2 Patienten), ETV6-NTRK3, TPM3-NTRK1, AFAP-NTRK1, AGTPBP1-NTRK2, KANK2-NTRK2, AGAP1-NTRK2, BCR-NTRK3, GKAP1-NTRK2, KANK-NTRK2, KCDT8-NTRK2, NTRK2-AGAP1, TNS3-NTRK2, sowie nicht bestimmt (bei je 1 Patient). Zum Zeitpunkt des Cut-off waren 14 der 18 aufgenommenen Patienten mit primären ZNS-Tumoren für das Ansprechen auswertbar. Ein komplettes Ansprechen wurde bei 1 Patienten beobachtet, eine partielle Remission bei 3 Patienten und bei 6 Patienten wurde eine stabile Erkrankung über mindestens 16 Wochen beobachtet. Die Gesamtansprechrate betrug 29% (95% Konfidenzintervall: 8%, 58%). Zum Zeitpunkt des Cut-off lag die Behandlungsdauer bei 0.03 bis 16.6 Monaten und wurde bei 13 von 18 Patienten fortgesetzt.
Unerwünschte Wirkungen
Die Sicherheit von Vitrakvi wurde an total 208 Patienten mit lokal fortgeschrittenen oder metastasierten soliden Tumoren (unabhängig vom NTRK-Genfusionsstatus) beurteilt, welche mindestens eine Dosis Vitrakvi in einer der drei klinischen Studien «LOXO-TRK-14001», «NAVIGATE» und «SCOUT» erhalten hatten. Die gesamte Sicherheitspopulation umfasste Patienten mit einem medianen Alter von 42 Jahren (28 Tage bis 82 Jahre), wobei 27% der Patienten pädiatrische Patienten (28 Tage bis 18 Jahre) waren. Die Mehrheit (82%) der Erwachsenen (ab 18 Jahren) erhielt 100 mg Vitrakvi zweimal täglich als Anfangsdosis. Die mediane Behandlungsdauer für die gesamte Sicherheitspopulation betrug 4,1 Monate (Spanne: 0,03 bis 40,7).
Die am häufigsten gemeldeten unerwünschten Wirkungen unter Vitrakvi waren Fatigue (36%), Schwindelgefühl (29%), Übelkeit (28%), Obstipation (27%), ALT erhöht (26%), AST erhöht (26%), Anämie (26%), Erbrechen (24%), Myalgie (16%), Gewichtszunahme (14%), Muskelschwäche (13%), Neutrophilenzahl erniedrigt (12%), Leukozytenzahl erniedrigt (11%). Die meisten unerwünschten Wirkungen waren von Grad 1 oder 2. Die höchsten für Vitrakvi gemeldeten Grade waren erniedrigte Neutrophilenzahl und erhöhte ALT (Grad 4) sowie Anämie, Fatigue, Schwindelgefühl, Gangstörung, Parästhesie, Übelkeit, Erbrechen, Obstipation, Myalgie, AST erhöht, Alkalische Phosphatase im Blut erhöht, Leukozytenzahl erniedrigt und Gewichtszunahme (Grad 3). Es wurden keine unerwünschten Wirkungen Grad 5 berichtet. Die meisten Ereignisse, die zu einer Dosisreduktion führten, traten innerhalb der ersten drei Behandlungsmonate auf. Zum dauerhaften Absetzen von Vitrakvi aufgrund behandlungsbedingter unerwünschter Wirkungen des Prüfpräparats kam es bei 2% der Patienten (ALT erhöht, AST erhöht, Muskelschwäche, Übelkeit, Enterokutanfistel und Amylase erhöht).
Kommentar
Es handelt sich um die zweite Zulassung, welche «tumor agnostic» erfolgt, d.h. dass nicht primär eine Indikation für eine einzelne organdefinierte Erkrankung, sondern für ein hochspezifisches Target verschiedener maligner Erkrankungen gleichzeitig erfolgt. Die erste solche Zulassung betraf Patienten mit einer «microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR)» Konstellation, welche von einer Pembrolizumab Therapie profitieren konnten.
Hier nun liegt ein für NTRK-Fusionsproteine hochselektives Medikament vor, welches eine klassische Orphan-Disease Situation betrifft. Es werden pro Jahr in Europa etwa 1000 solche Neuerkrankungen erwartet. Die aktuell bereits ersichtliche Wirksamkeit ist klinisch bedeutsam und ohne Alternative für diese Patientengruppen. Die Nebenwirkungen sind nach bisheriger Erfahrung moderat. Allerdings ist angesichts der Seltenheit dieser Tumorerkrankungen die Datenlage noch sehr schmal und entsprechend laufen die Studien weiter. Damit es gelingt, die Sicherheit hoch zu halten, sollten solche Therapien nur an Zentren zur Anwendung kommen, wo solche Therapien regelmässig zur Anwendung kommen, damit eine genügende Erfahrung zur weiteren Optimierung erlangt werden kann. Die seit Januar 2020 auch in der Schweiz nun mögliche zeitlich befristete Zulassung nach Art. 9a des Heilmittelgesetzes wird in den folgenden Jahren zeigen müssen, ob die aktuelle Datenlage durch Langzeiterfahrungen bestätigt wird und somit diese frühe Zulassung von Vitrakvi aufrechterhalten werden kann.
Copyright bei Aerzteverlag medinfo AG
Der Autor hat keine Interessenskonflikte
im Zusammenhang mit diesem Beitrag deklariert.
Swissmedic genehmigte Fachinformation zu Vitrakvi Kapsel 2020