
En décembre 2020, Swissmedic a autorisé le médicament orphelin Trikafta®, une combinaison hautement efficace de modulateurs de la protéine CFTR (Cystic fibrosis transmembrane conductance regulator) pour le traitement de la mucoviscidose chez les patients porteurs d’ au moins une mutation F508del du gène CFTR, mutation la plus fréquente dans la mucoviscidose. Les principes actifs sont l’ élexacaftor, le tézacaftor et l’ ivacaftor. Actuellement, plus de 85% de patients avec mucoviscidose sont éligibles pour un traitement par modulateurs du CFTR, qui améliore la fonction pulmonaire et la qualité de vie et diminue les exacerbations respiratoires.
In December 2020, Swissmedic approved the drug Trikafta®, a highly effective combination of modulators of the CFTR protein (Cystic fibrosis transmembrane conductance regulator), for the treatment of cystic fibrosis (CF) in patients with at least one F508del mutation of the CFTR gene (the most common mutation in CF). The active substances are elexacaftor, tezacaftor and ivacaftor. Currently, more than 85% of people with CF are eligible for CFTR modulator therapy, which has been shown to improve respiratory function and quality of life and reduce pulmonary exacerbations.
Key Words: cystic fibrosis, modulators of the CFTR, elexacaftor, tezacaftor, ivacaftor
La mucoviscidose
La mucoviscidose est une maladie monogénique potentiellement grave, à transmission autosomique récessive, touchant plus de 100’ 000 personnes dans le monde (1, 2). Elle est due à des mutations sur le gène CFTR entraînant une altération de la synthèse ou de la fonction de la protéine CFTR (Cystic Fibrosis Transmembrane conductance Regulator, ci-après pCFTR). Depuis la découverte initiale de la mutation la plus courante (F508del), plus de 2000 variants du gène CFTR ont été décrits (1, 2), dont environ 400 sont considérés associés à la mucoviscidose. Ils sont répertoriés dans les classes I à VI, en fonction des de leur impact sur la production et la fonction de la pCFTR (1, 2). La pCFTR est un canal transmembranaire de chlorure qui se trouve sur la face apicale des épithéliums sécrétoires, plus particulièrement les glandes sudoripares, les voies respiratoires, le tractus gastro-intestinal, le pancréas et les canaux déférents (1, 2). Elle régule l’ équilibre en sel et en eau à la surface des cellules et un dysfonctionnement de cette protéine résulte en des sécrétions et mucus visqueux. Les manifestations cliniques de la maladie sont multisystémiques et se déclarent à des âges variables selon le dysfonctionnement de la pCFTR (3, 4). Au niveau pulmonaire, la mucoviscidose est caractérisée par un trouble sévère de la clairance mucociliaire, une inflammation et une colonisation bactérienne chronique des voies respiratoires supérieures et inférieures, se compliquant de bronchectasies et d’ un déclin progressif de la fonction pulmonaire. L’ insuffisance pancréatique exocrine, avec malabsorption des lipides et des vitamines liposolubles, est très fréquente, ainsi que la constipation. Le diabète lié à la mucoviscidose concerne 30 % des patients de plus de 18 ans et sa prévalence augmente avec l’ âge, alors que la cirrhose biliaire est beaucoup plus rare (3, 4). La prise en charge de la mucoviscidose a longtemps reposé sur un traitement symptomatique contraignant, mais qui a prolongé considérablement l’ espérance de vie des patients. Depuis les années 2010, une nouvelle classe de médicaments est disponible, les modulateurs de la pCFTR permettant de restaurer l’ activité de celle-ci (1).
Les modulateurs de la protéine du CFTR
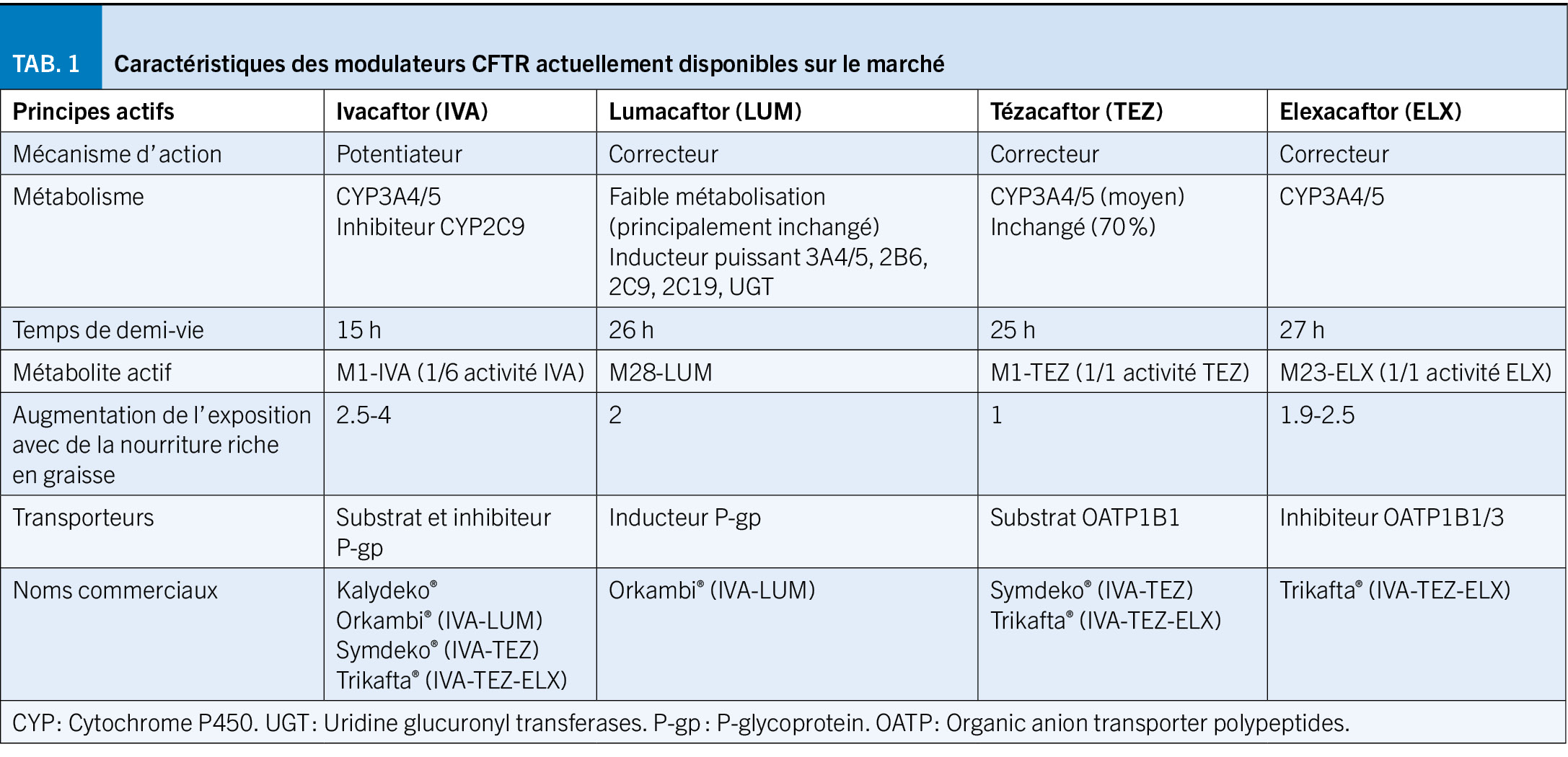
Contrairement aux thérapies symptomatiques axées sur le traitement des complications de la mucoviscidose, les modulateurs de la pCFTR sont des petites molécules qui agissent à l’ origine du problème (Figure 1). Il existe deux types de modulateurs : les potentiateurs (ivacaftor) améliorent la fonction de la pCFTR, favorisant le temps d’ ouverture des canaux chlorure. Les correcteurs (lumacaftor, tézacaftor et élexacaftor) stabilisent la pCFTR et facilitent son transport vers la membrane cellulaire (1). Quatre médicaments en mono-, bi- ou trithérapie existent actuellement sur le marché dont quelques caractéristiques sont décrites dans le Tableau 1.
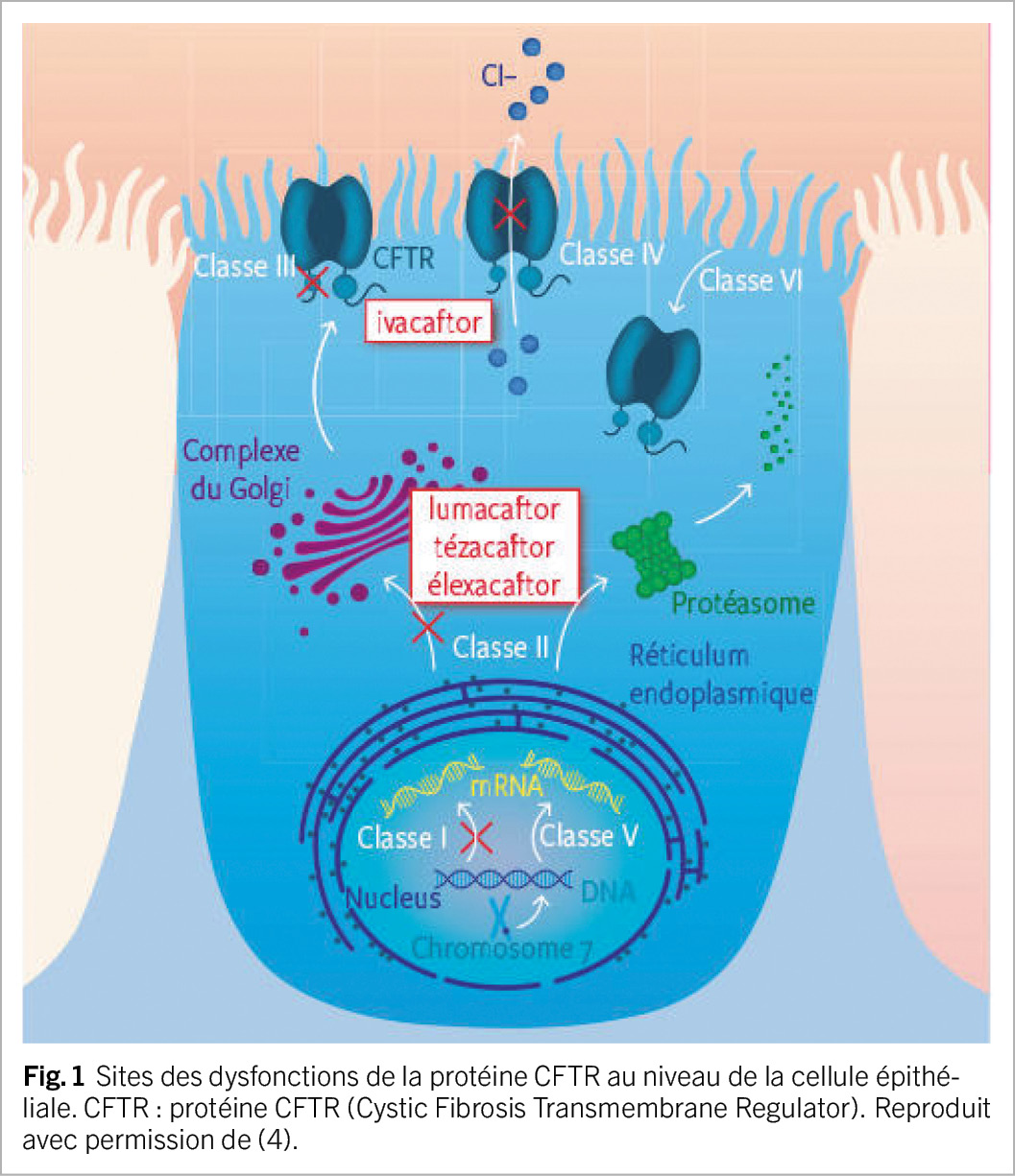
Efficacité et sécurité des modulateurs
En 2019, 2 études pivots ont été publiées concernant la trithérapie élexacaftor/tézacaftor/ivacaftor (Trikafta®) chez 113 patients homozygotes F508del (5) et 403 patients hétérozygotes composites F508del/variant à fonction minimale non corrigeable par les associations de modulateurs précédentes (6). La première étude n’ a porté que sur 4 semaines et le groupe témoin était constitué de patients traités par ivacaftor/tézacaftor. L’ ajout de l’ élexacaftor a permis un gain supplémentaire de 10,0 % du volume expiratoire maximal en une seconde (VEMS) et une réduction du chlorure sudoral de 45 mmol/l (p < 0,0001). La deuxième étude a entraîné une amélioration du VEMS de 13,9 % à 24 semaines, une diminution de 63 % du taux d’ exacerbations pulmonaires, une amélioration du score CFQ-R (score de qualité de vie) de 20,2 points et une baisse de chlorure dans la sueur de 42 mmol/l (p<0,001 pour toutes les comparaisons). Ces résultats ont été confirmés par des études prospectives de vie réelle qui ont également permis d’ étendre l’ enregistrement aux enfants dès 6 ans et très récemment dès 2 ans par la FDA (7, 8).
D’ un point de vue de la toxicité, les modulateurs sont relativement bien tolérés (9-11). La combinaison ivacaftor/lumacaftor présente cependant des taux plus élevés d’ effets indésirables respiratoires qui semblent dus au lumacaftor (10). Une augmentation transitoire de la toux/bronchorrhée dans les 48 heures après le début du traitement est la conséquence de l’ amélioration de la clearance des sécrétions et diminue par la suite. Les autres effets indésirables se manifestent surtout au niveau gastro-intestinal (diarrhées, nausées, douleurs abdominales) et hépatique (augmentation des transaminases) (9-11). Des allergies de type rash ou d’ hypersensibilité ont été rarement rapportées. Un signal d’ une atteinte de la santé mentale (notamment des dépressions ou troubles neuropsychiatriques) a été identifié sur la base de quelques études de cohorte et de rapport de cas (9), bien qu’ un lien de causalité avec les modulateurs du CFTR n’ aie pas pu être formellement établi.
Considérations pharmacocinétiques
Les modulateurs de la pCFTR sont éliminés majoritairement par les cytochromes (CYP) 3A4/5 (Tableau 1) et l’ ivacaftor et l’ élexacaftor peuvent aussi être substrats, inhibiteurs ou inducteurs des P-gp et/ou des OATP1B1/3. De ce fait, ils présentent un risque important d’ interactions médicamenteuses, en particulier avec les inducteurs ou les inhibiteurs des CYP3A4/5. Une diminution marquée de l’ exposition (89 %) et une augmentation de 15 fois des concentrations d’ ivacaftor en co-administration avec la rifampicine et l’ itraconazole, respectivement, ont été observées (11). Des modifications plus faibles toutefois significatives sont attendues avec les inhibiteurs ou inducteurs modérés des CYP3A4/5 (p.ex. rifabutine ou fluconazole). Bien qu’ aucune interaction médicamenteuse entre le Trikafta® et les inducteurs de l’ OATP1B1/3 n’ ait été rapportée, le gemfibrozil et la ciclosporine, inhibiteurs de l’ OATP1B1/3, pourraient théoriquement entraîner une augmentation des concentrations sériques de Trikafta® (12). A noter également que l’ ivacaftor, inhibiteur des CYP2C9, peut augmenter l’ effet de certains médicaments substrat de cette enzyme (acénocoumarol, glibenclamide) et une prudence et une surveillance adaptée est préconisée en cas d’ administration de médicaments substrats des OATP1B1 tels que les statines, le glibenclamide ou le répaglinide. La bilirubine étant un substrat d’ OATP1B1 et d’ OATP1B3, de légères augmentations du taux moyen de bilirubine totale peuvent survenir (10, 11).
Une importante variabilité des concentrations plasmatiques a été reportée entre patients traités par Trikafta®, associée à certains facteurs tels l’ âge et le poids chez les enfants, la nourriture riche en graisse augmentant significativement l’ exposition à ces médicaments, les co-médications à risque d’ interactions et les altérations de l’ élimination en cas d’ insuffisance hépatique ou rénale. L’ impact de ces variations pharmacocinétiques sur la réponse et la tolérance à ces médicaments est toutefois encore largement inconnu. Le suivi thérapeutique par mesure des taux plasmatiques sera potentiellement utile afin d’ individualiser les posologies de ces médicaments (13).
Points de vigilance en pratique
Les doses de Trikafta® doivent être prises à environ 12 heures d’ intervalle avec un repas riche en graisses. En cas d’ insuffisance hépatique modérée, la posologie doit être réduite (11).
La prescription concomitante d’ inducteurs puissants des CYP3A4/5 est contre-indiquée en raison du risque de perte d’ efficacité du médicament. Une réduction posologique est recommandée avec les inhibiteurs forts et faibles des CYP3A4/5 pour diminuer le risque d’ effets indésirables. La prudence et une surveillance appropriée s’ imposent avec les médicaments substrats du CYP2C9, de la P-gp et des OATP1B1 et OATP1B3 co-administrés avec le Trikafta® en raison de l’ augmentation possible de l’ exposition à ces médicaments.
Il est recommandé de contrôler les taux de transaminases (ALAT et ASAT) et de bilirubine totale chez tous les patients avant l’ instauration du traitement, tous les trois mois durant la première année, puis au moins une fois par an (11). En raison du risque de détérioration respiratoire sévère après l’ interruption de ce traitement, un soutien à l’ adhésion thérapeutique et la bonne connaissance des enjeux par le patient et le personnel soignant est nécessaire (14).
Finalement, le coût d’ un emballage de 84 comprimés (28 jours) de Trikafta® est de 17’ 516.15 CHF et il faut l’ accord de prise en charge des assureurs avec une évaluation préalable par le médecin-conseil de l’ assurance.
Conclusion
Les modulateurs de la pCFTR ont révolutionné la prise en charge des patients souffrant de mucoviscidose, entraînant une amélioration significative de la fonction pulmonaire et de la qualité de vie, ainsi qu’ une réduction du risque d’ exacerbations pulmonaires. Comme pour tout nouveau traitement, il subsiste néanmoins des inconnues sur l’ efficacité, notamment extrapulmonaire, à long terme et les effets indésirables à long terme et certaines précautions doivent être prises afin de minimiser les risques de toxicité. D’ autres principes actifs sont en cours d’ investigation et seront mis sur le marché ces prochaines années (15).
Copyright Aerzteverlag medinfo AG
Service de pharmacie, Centre Hospitalier Universitaire Vaudois et
Université de Lausanne
ermindo.di-paolo@chuv.ch
Unité de mucoviscidose adulte, Service de Pneumologie,
Centre Hospitalier Universitaire Vaudois et Université de Lausanne
georgia.mitropoulou@chuv.ch
Centre de Recherche et d’ Innovation en Sciences Pharmaceutiques
Cliniques
Centre Hospitalier Universitaire et Université de Lausanne
Rue du Bugnon 17
1011 Lausanne
Chantal.Csajka@chuv.ch
Les auteurs n’ont pas déclaré de conflits d’ intérêts en rapport avec cet article.
1. Shteinberg M, Haq IJ, Polineni D, et al. Cystic fibrosis. Lancet 2021;397:2195-2211.
2. Bell SC, Mall MA, Gutierrez H, et al. The future of cystic fibrosis care: a global perspective. Lancet Respir Med 2020; 8:65-124.
3. Koutsokera A, Sauty A et al. Swiss recommendations for adult cystic fibrosis care. https://www.revmed.ch/guidelines/swiss-recommendations-for-adult-cystic-fibrosis-care (dernier accès 20.02.2023).
4. Sauty A, Plojoux J, Mornand A, et al. Révolution dans le traitement de la mucoviscidose. Rev Med Suisse 2020; 16:1229-35.
5. Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet 2019;394:1940-8.
6. Middleton PG, Mall MA, Drevínek P, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med 2019;381:1809-19.
7. Trikafta® Swiss Public Assessment Report. Swissmedicinfo. 30 March 2022.
8. Nichols DP, Paynter AC, Heltshe SL, et al. Clinical effectiveness of Elexacaftor/Tezacaftor/Ivacaftor in people with cystic fibrosis: a clinical trial. Am J Respir Crit Care Med 2022;205:529-39.
9. Dagenais RVE, Su VCH, Quon BS. Real-world safety of CFTR modulators in the treatment of cystic fibrosis: a systematic review. J Clin Med 2020;10:23. doi: 10.3390/jcm10010023.
10. Gramegna A, Contarini M, Aliberti S, et al. From ivacaftor to triple combination: a systematic review of efficacy and safety of CFTR modulators in people with cystic fibrosis. Int J Mol Sci 2020 ;21:5882. doi: 10.3390/ijms21165882.
11. https://www.swissmedicinfo.ch, monographie Trikafta® (dernier accès 20.02.2023).
12. Purkayastha D, Agtarap K, Wong K, et al. Drug-drug interactions with CFTR modulator therapy in cystic fibrosis: focus on Trikafta®/Kaftrio®. J Cyst Fibros 2023. doi: 10.1016/j.jcf.2023.01.005.
13. Choong E, Sauty A, Koutsokera A, et al. Therapeutic drug monitoring of ivacaftor, lumacaftor, tezacaftor, and elexacaftor in cystic fibrosis: where are we now? Pharmaceutics 2022;14:1674. doi: 10.3390/pharmaceutics14081674.
14. Mitropoulou G, Balmpouzis Z, Plojoux, J, et al. Effects of elexacaftor−tezacaftor−ivacaftor discontinuation in cystic fibrosis. Respir Med Res 2022;82:100972. doi: 10.1016/j.resmer.2022.100972.
15. https://apps.cff.org/trials/pipeline (dernier accès 20.02.2023).



la gazette médicale
- Vol. 12
- Ausgabe 6
- Oktober 2023