
La tendance clinique constitutionnelle aux hémorragies est une réalité. Celle-ci repose à la fois sur des modifications acquises et héréditaires de la coagulation. Les jeunes, qui présentent moins de risques d’exposition que les personnes plus âgées, ne se font pas immédiatement remarquer sur le plan clinique. C’est pourquoi un diagnostic de laboratoire approfondi et précoce est très utile et judicieux.
Abstract: The constitutional clinical bleeding tendency is a reality. This is based on both acquired and hereditary changes in coagulation. Young people with fewer exposure risks compared to the elderly do not immediately stand out clinically. Therefore, thorough early laboratory diagnosis is very helpful and useful.
Key Words: Coagulation, bleedings, thrombocytes
Les mécanismes hémostatiques constituent un système biologique complexe où diverses enzymes de coagulation solubles interagissent avec les structures cellulaires dans une cascade d’ activation mutuelle. Le système reste « piégé « dans le réseau vasculaire et se trouve principalement dans un « état de repos «. Lors d’ une lésion mécanique ou toxique de la paroi vasculaire, une perte de sang se produit en parallèle et déclenche la cascade des interactions de la coagulation. Ceci conduit inévitablement à la formation du thrombus hémostatique. Les partenaires de ces interactions sont aujourd’ hui connus, tant dans leur structure que dans leur fonction (fig. 1).
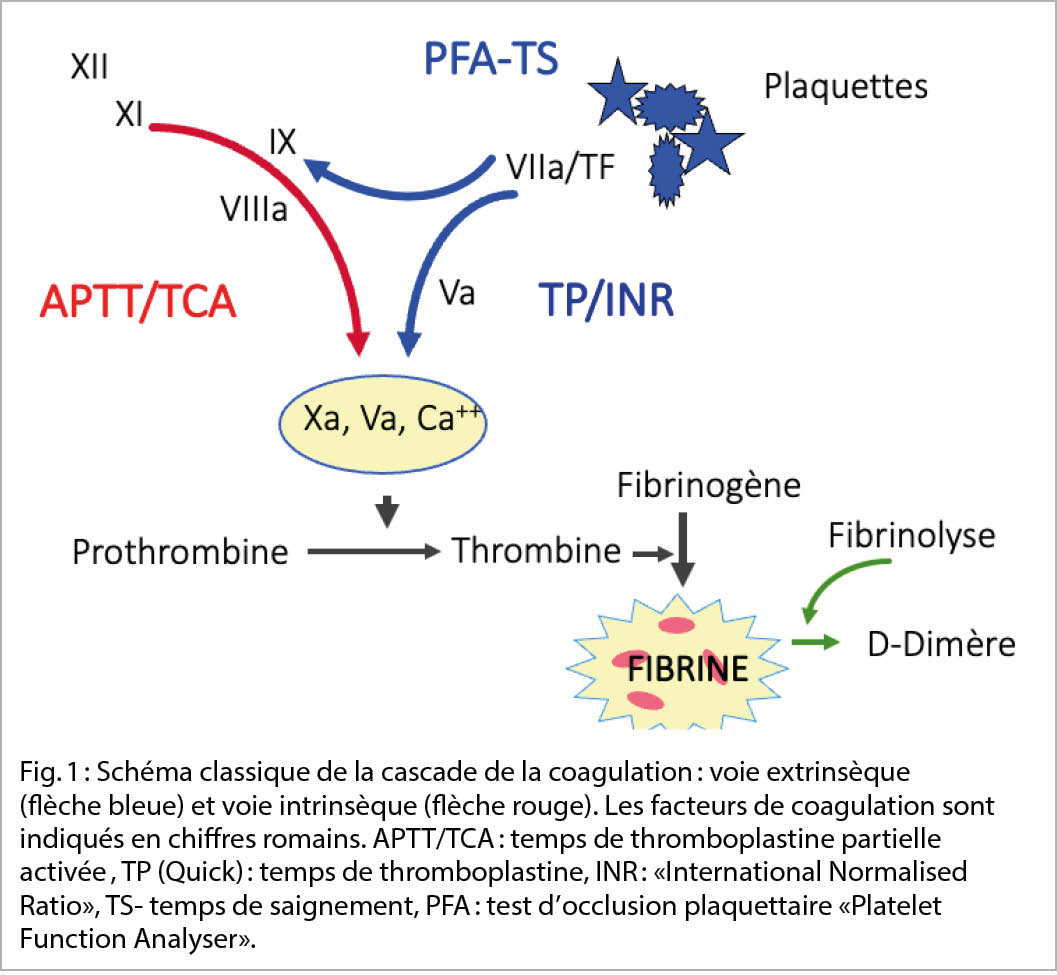
En cas de déficit d’ un facteur de la coagulation ou d’ une inhibition de sa fonction, la voie physiologique de la coagulation subit un ralentissement qui se traduit cliniquement par une tendance aux saignements.
Les personnes avec tendance aux saignements peuvent présenter comme signes cliniques des pétéchies (taches rouges de la peau de taille d’ une tête d’ épingle), des ecchymoses (taches cutanées de la taille d’ une pièce de monnaie), des suffusions (saignements diffus sur une plus grande surface) ou les hématomes (saignements délimités). Ces présentations cliniques peuvent parfois orienter vers la cause de l’ hémorragie.
Par exemple, les anomalies vasculaires se manifestent généralement par des hématomes, des hémorragies cutanées ou gastro-intestinales. Les anomalies plaquettaires provoquent des pétéchies, des hémorragies gingivales ou nasales, ainsi que des hémorragies gastro-intestinales ou du système nerveux central. Les troubles de la coagulation plasmatique se manifestent souvent par des hématomes dans les ligaments musculaires, des hémorragies cutanées ou intra-articulaires, ainsi que des hémorragies après un traumatisme.
L’ anamnèse détaillée et les antécédents de la personne, en plus de l’ examen clinique et des analyses ciblées de laboratoire permettent presque toujours d’ identifier avec précision le trouble de la coagulation.
Clarification première
Antécédents médicaux
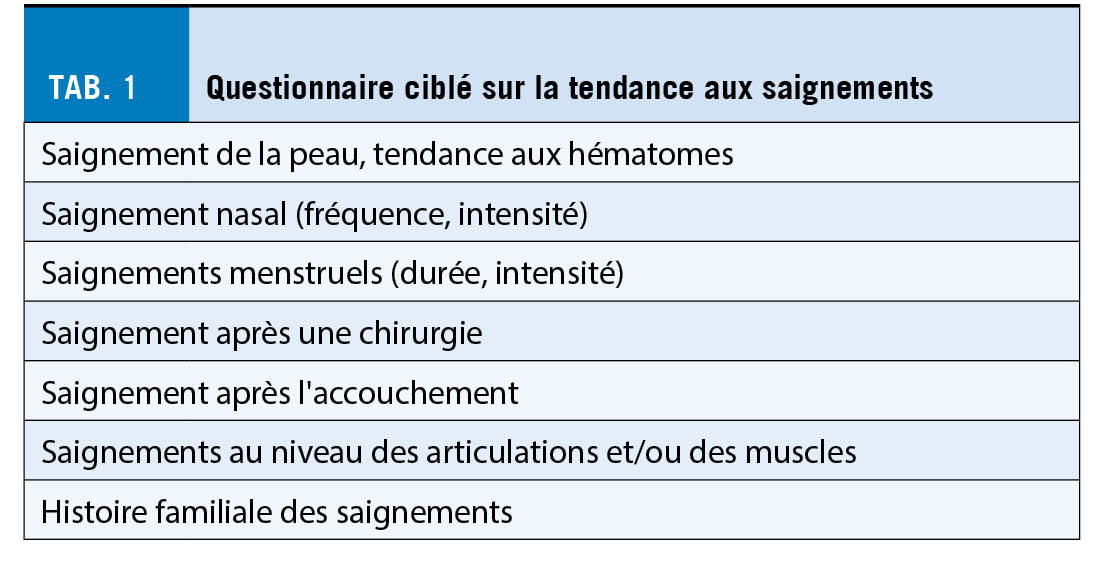
Un historique spécifique et détaillé des saignements est très utile. Comme les patients omettent souvent des épisodes isolés de saignement, ils doivent être interrogés de manière ciblée avant toute intervention diagnostique ou thérapeutique (tab. 1). Par exemple, des saignements menstruels abondants depuis la ménarche nécessitent une clarification pour la maladie de von Willebrand. Plus le taux du facteur de von Willebrand est faible, plus la tendance aux saignements est forte. Des questionnaires structurés au sens d’ un score, comme le score de détection des saignements BAT-ISTH de la Société Internationale sur la Thrombose et l’ Hémostase ISTH, peuvent également être utiles.
Constatations objectives
Dans le cas de déficits légers des facteurs de la coagulation, il n’ y a pas de saignements spontanés. Ces derniers peuvent être provoqués dans le contexte d’ un traumatisme, une chirurgie ou une anticoagulation. Dans le cas de déficits modérés ou graves, les saignements peuvent également se produire spontanément. Par exemple, chez les patients âgés souffrant d’ une hémophilie sévère, on observe des atteintes impressionnantes de l’ appareil locomoteur (arthropathie hémophilique, atrophie musculaire, déformations, contractures, etc.). Ces complications, sont en général absentes chez les patients hémophiles de plus jeune âge en raison de la substitution prophylactique du facteur déficient. En plus du tableau classique des manifestations hémorragiques, les personnes avec une anamnèse personnelle et familiale de tendance aux saignements doivent également être considérées à risque hémorragique jusqu’ à preuve du contraire.
Analyses de laboratoire
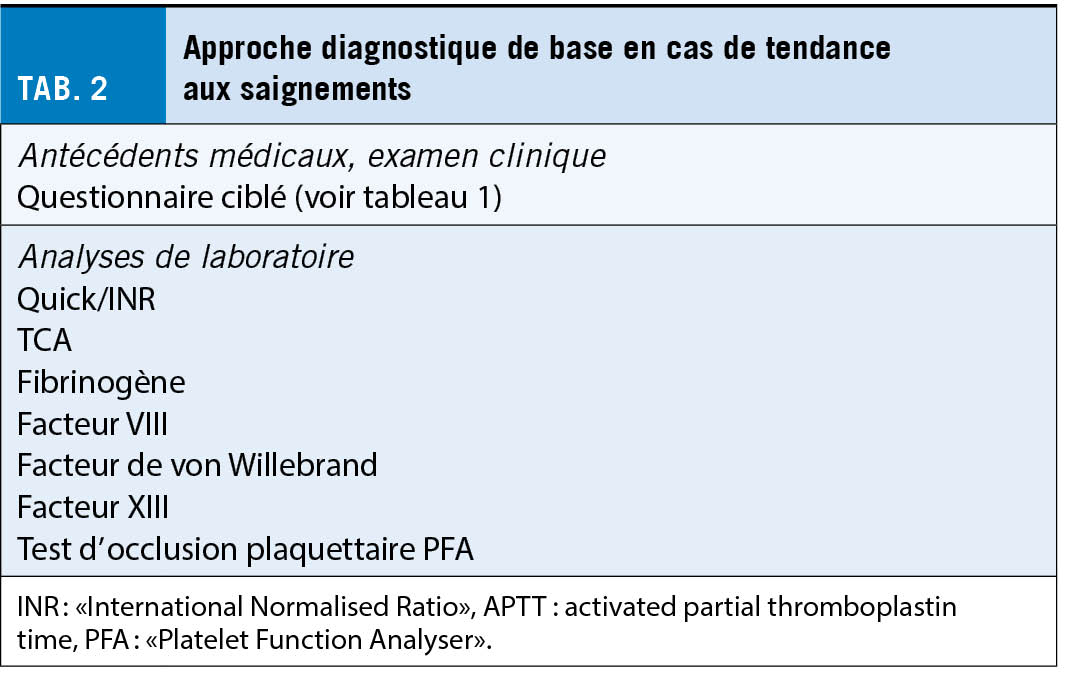
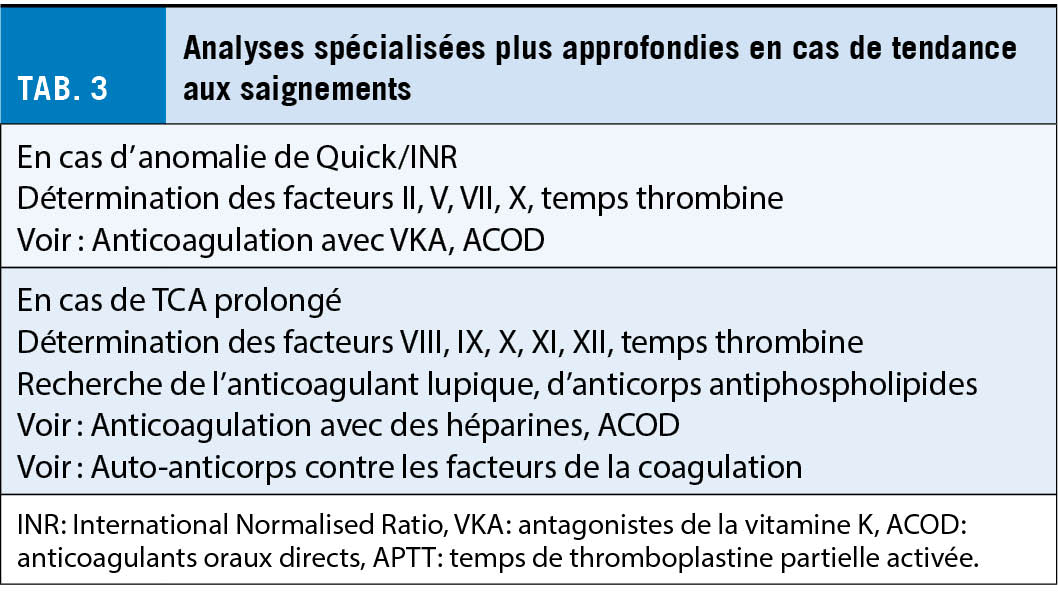
Si l’ on suspecte une tendance héréditaire ou acquise hémorragique, un groupe d’ analyses de base doit être prescrit. Cela comprend une formule sanguine et les tests globaux de coagulation et la fonction plaquettaire (test d’ occlusion plaquettaire PFA). En outre, le facteur de von Willebrand et le facteur XIII sont également à prévoir car ils ne sont pas détectés par les tests globaux (tab. 2). En fonction des résultats des analyses de base, les investigations peuvent être complétées par des analyses plus approfondies, telles que la détermination spécifique des facteurs de coagulation ou l’ analyse de la fonction plaquettaire au moyen de l’ agrégation plaquettaire et/ou de l’ immunophénotypage (tab. 3).
Analyses spécialisées
Étude ciblée de l’ agrégation plaquettaire
L’ agrégation plaquettaire permet d’ étudier et de classer les troubles de la fonction plaquettaire et met en évidence les défauts des récepteurs membranaires des plaquettes. La capacité de stimulation plaquettaire par divers agonistes naturels (ADP, collagène, adrénaline, ristocétine, acide arachidonique, thrombine (TRAP) et agoniste du récepteur de la thromboxane) in vitro est déterminée par turbidimétrie.
Caractérisation ciblée des récepteurs membranaires des plaquettes (immunophénotypage)
Cette analyse est prescrite si l’ on soupçonne de thrombopathies héréditaires et aide à diagnostiquer et à caractériser les défauts des récepteurs membranaires des plaquettes. Des anticorps monoclonaux et la méthode de cytométrie en flux sont utilisés pour déterminer de manière semi-quantitative la densité des récepteurs à la surface des plaquettes avant et après l’ activation plaquettaire in-vitro.
Test des multimères du facteur de von Willebrand (VWF-MM)
Le syndrome de von Willebrand est le trouble héréditaire de la coagulation le plus fréquent (incidence de 1 / 200-300) et est diagnostiqué par le dosage du facteur de von Willebrand (activité et antigène) et du facteur VIII. Selon les résultats de ces analyses, la clarification est complétée par l’ analyse du VWF-MM basé sur la caractérisation qualitative et quantitative des multimères de vWF circulants au moyen d’ électrophorèse sur gel.
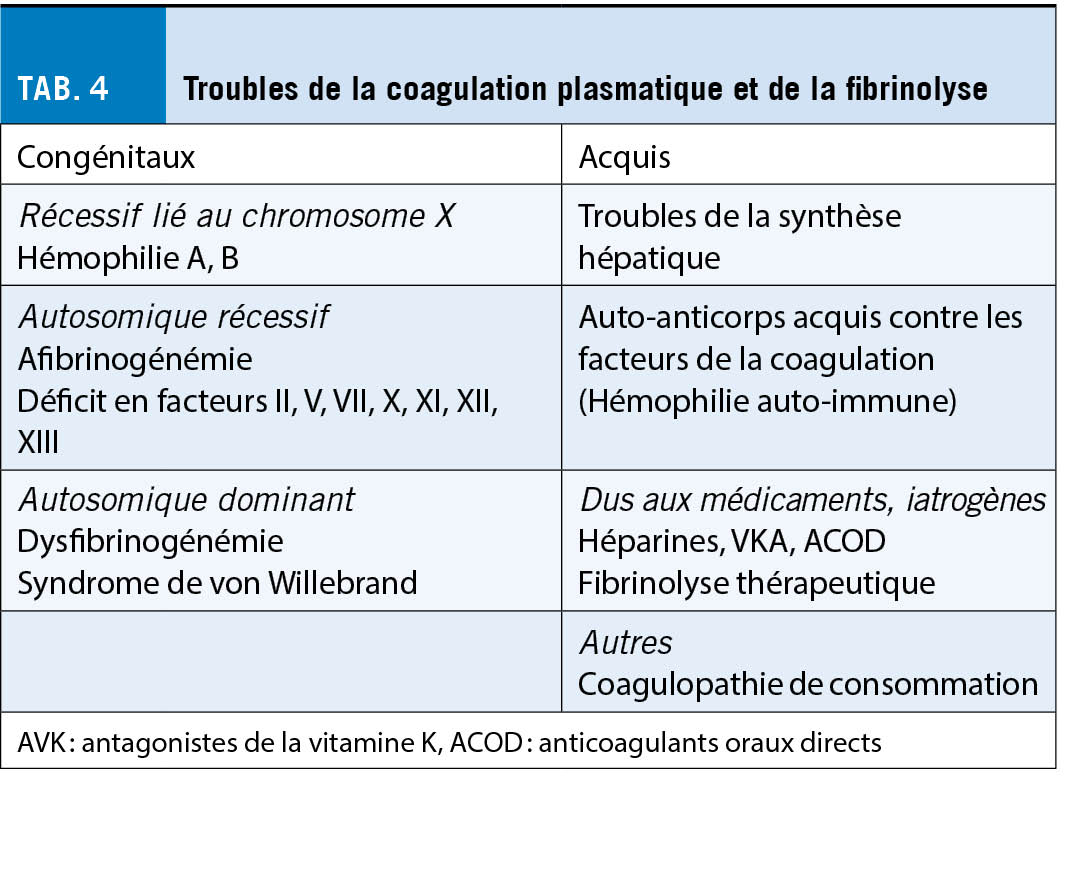
Présentation clinique et approche diagnostique en cas de tendance aux saignements (tableau 4)
Hémophilie A et B
L’ hémophilie est une diminution héréditaire des facteurs de la coagulation VIII ou IX, liée au chromosome X. Les hommes sont touchés, les femmes sont conductrices. Les hémorragies articulaires et musculaires sont typiques. Une tendance aux saignements massifs est à prévoir en cas de traumatisme ou de chirurgie.
Dans les cas d’ hémophilie A ou B sévère et modérée, le TCA est nettement allongé, alors que dans les cas d’ hémophilie légère, il n’ est que légèrement allongé ou reste dans l’ intervalle de la norme. En cas de suspicion basée sur l’ anamnèse, le TCA normal ne permet donc pas d’ exclure une hémophilie légère. La détermination spécifique du facteur VIII ou IX permet d’ identifier précisément le défaut.
Le syndrome de von Willebrand
Il s’ agit d’ un trouble héréditaire autosomique du facteur de von Willebrand (vWF) avec une variété d’ anomalies du gène du vWF. Le vWF est important pour l’ adhésion des plaquettes à l’ intima du vaisseau abimé et pour leur activation. Il stabilise également le facteur VIII, avec lequel il se lie en complexe et circule dans le plasma. Les formes bénignes du syndrome de von Willebrand sont prédominantes, les patients ne saignent que lors d’ un challenge de l’ hémostase et ont généralement un temps de saignement normal. Tous les types de syndrome de von Willebrand confondus, on estime qu’ ils touchent près de 1 % de la population. La forme la plus grave (type 3) a une incidence de 1 par million.
Le diagnostic repose sur la détermination quantitative du vWF (activité et antigène) et de l’ activité du facteur VIII. La classification des types de syndrome de von Willebrand est en constante évolution, bien que la classification mentionnée ci-dessous ait été jusqu’ à présent suffisante pour répondre aux besoins cliniques.
Type 1 : Réduction concordante du vWF fonctionnel et antigénique à moins de 50 % de la norme (habituellement 5-30 %) et du facteur VIII de 50 % ou moins. Transmis de manière autosomique dominante, ce type représente environ 70 % des patients. Attention : les personnes du groupe sanguin O ont des taux de vWF physiologiquement plus faibles sans tendance clinique aux saignements. Le cut-off est fixé à 35 % au lieu de 50 %.
Type 2A : Diminution de l’ activité du vWF accompagnée d’ un antigène du vWF normal ou seulement légèrement diminué (rapport activité du vWF/antigène du vWF < 0,7). L’ analyse des multimères montre une réduction des chaînes de poids moléculaire élevé particulièrement actives sur le plan fonctionnel. Facteur VIII normal ou légèrement réduit. Transmission autosomique dominante (parfois récessive).
Type 2B : Comme le type 2A, mais accompagné d’ une augmentation paradoxale de l’ agrégation induite par la ristocétine du plasma riche en plaquettes du patient, en raison de l’ affinité accrue des chaînes anormales de vWF pour le récepteur GP Ib/V/IX (récepteur du facteur de von Willebrand) du patient. Dans l’ analyse multimérique du vWF, réduction des grandes et moyennes chaînes du vWF. La stimulation de la libération de vWF anormal entraîne une thrombopénie. Transmission autosomique dominante.
Type 2M : Faible affinité plaquettaire des multimères de vWF. Contrairement aux types 2A et 2B, les grands multimères sont également présents, mais leur fonction est altérée, ce qui se manifeste par une fonction réduite du vWF avec un antigène vWF encore normal et une électrophorèse normale des multimères du vWF (mais une forme anormale des triplets de bandes des multimères). Transmission autosomique dominante.
Type 2N, vWF type Normandie : vWF avec un site de liaison anormal pour le facteur VIII, qui est rapidement éliminé en raison de l’ absence de liaison avec le FvW par ailleurs quantitativement et qualitativement normal. Le tableau clinique d’ une hémophilie A légère est présent (également chez les femmes) avec un VIII autour de 5-30 %. Transmission autosomique récessive.
Type 3 : activité et antigénique indétectable. Le facteur VIII est également réduit en raison de l’ absence de protéine de transport dans le plasma. Transmission autosomique récessive. Temps de saignement considérablement prolongé.
Troubles de la coagulation rares
Les très rares déficits isolés héréditaires des facteurs de la coagulation se manifestent par des tests globaux anormaux : II, V et X par un Quick/INR anormal et un TCA anormal, VII par un Quick / INR anormal avec un TCA normal et les protéines de la phase de contact XII et le facteur XI par un allongement isolé du TCA. Dans l’ afibrinogénémie, la formation de caillots est absente dans tous les tests globaux. Le diagnostic est établi par la détermination du facteur spécifique. Le déficit en facteur XII n’ est pas associé à une tendance aux saignements. De même, le déficit en facteur VII avec un taux résiduel supérieur à 10 % de la norme est asymptomatique.
Dysfonctionnement plaquettaire héréditaire
Les hémorragies les plus graves sont observées dans les troubles plaquettaires classiques et rares, telles que la thrombasthénie de Glanzmann (défaut du récepteur du fibrinogène GP IIb / IIIa) et le syndrome de Bernard-Soulier (défaut du récepteur du vWF GP Ib / V / IX). Le temps de saignement est généralement anormal, les analyses d’ agrégation plaquettaire et la caractérisation des récepteurs de la surface plaquettaire confirment le défaut.
Il existe également d’ anomalies plaquettaires associées à des défauts d’ autres récepteurs de surface, la transduction du signal, la densité des granules alpha et le métabolisme de l’ acide arachidonique.
Dre Leda Leoncini, Dr Mario Uhr
SYNLAB Suisse SA, Via Pianon 7, 6934 Bioggio
(ledaleoncini@ticino.com, mario.uhr@synlab.com)
Dre Yordanka Tirefort
SYNLAB Suisse SA, Ch. d’Entre-Bois 21, 1018 Lausanne
(yordanka.tirefort@synlab.com)
Pr Dr Dimitrios Tsakiris
SYNLAB Suisse SA, Alpenquai 14, 6002 Luzern
(dimitrios.tsakiris@synlab.com)
Article traduit de «der informierte arzt» 12_2021.
Copyright Aerzteverlag medinfo
SYNLAB Suisse SA
Via Pianon 7
6934 Bioggio
leda.leoncini@synlab.com
Klinik für Hämatologie
Hämatologische Diagnostik Labormedizin
Universitätsspital Basel und Blutspendezentrum beider Basel SRK
Petersgraben 4
4031 Basel
Les auteurs déclarent n’ avoir aucun conflit d’ intérêts en rapport avec cet article.
1. Hayward CPM. How I investigate for bleeding disorders. Int J Lab Hematol. 2018 May;40 Suppl 1:6-14. doi: 10.1111/ijlh.12822. PMID: 29741250.
2. Boender J, Kruip MJ, Leebeek FW. A diagnostic approach to mild bleeding disorders. J Thromb Haemost. 2016 Aug;14(8):1507-16. doi: 10.1111/jth.13368. Epub 2016 Jun 27. PMID: 27208505.
3. Hayward CPM, Moffat KA, Brunet J, Carlino SA, Plumhoff E, Meijer P, ZehnderJL. Update on diagnostic testing for platelet function disorders: What is practical and useful? Int J Lab Hematol. 2019 May;41 Suppl 1:26-32. doi: 10.1111/ijlh.12995. PMID: 31069975.
4. James PD. Women and bleeding disorders: diagnostic challenges. Hematology AmSoc Hematol Educ Program. 2020 Dec 4;2020(1):547-552. doi:10.1182/hematology.2020000140. PMID: 33275722; PMCID: PMC7727580.
5. Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, Neunert C, Lillicrap D; ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010 Sep;8(9):2063-5. doi: 10.1111/j.1538-7836.2010.03975.x. PMID: 20626619


la gazette médicale
- Vol. 11
- Ausgabe 1
- Januar 2022