
Die neuroendokrinen Neoplasien (NEN) entstehen aus Zellen des diffusen neuroendokrinen Systems und können deshalb in den verschiedensten Organen auftreten (1,2). Charakteristisch ist die langsame Wachstumsrate und die Expression neuroendokriner Marker (wie Synaptophysin, Chromogranin A, neuronspezifische Enolase NSE oder CD56). Ausserdem haben sie die Fähigkeit, eine Vielzahl unterschiedlicher Hormone, Peptide oder biogene Amine zu sezernieren.
The neuroendocrine neoplasms (NEN) arise from cells of the diffuse neuroendocrine system and can therefore occur in a wide variety of organs (1,2). They are characterized by a slow growth rate and expression of neuroendocrine markers (such as synaptophysin, chromogranin A, neuron-specific enolase NSE or CD56). Furthermore, they have the ability to secrete a variety of different hormones, peptides or biogenic amines.
Key Words: Neuroendocrine neoplasms (NEN), neuroendocrine tumors (NET)
Trotz dieser Gemeinsamkeiten handelt es sich um eine sehr heterogene Tumorentität mit häufig unspezifischen Symptomen und einer äusserst variablen klinischen Präsentation. Dies führt in vielen Fällen zu langen diagnostischen Irrwegen mit belastenden Unsicherheiten für die Patienten und Verzögerungen in der Einleitung der korrekten Therapieschritte (3).
Epidemiologie
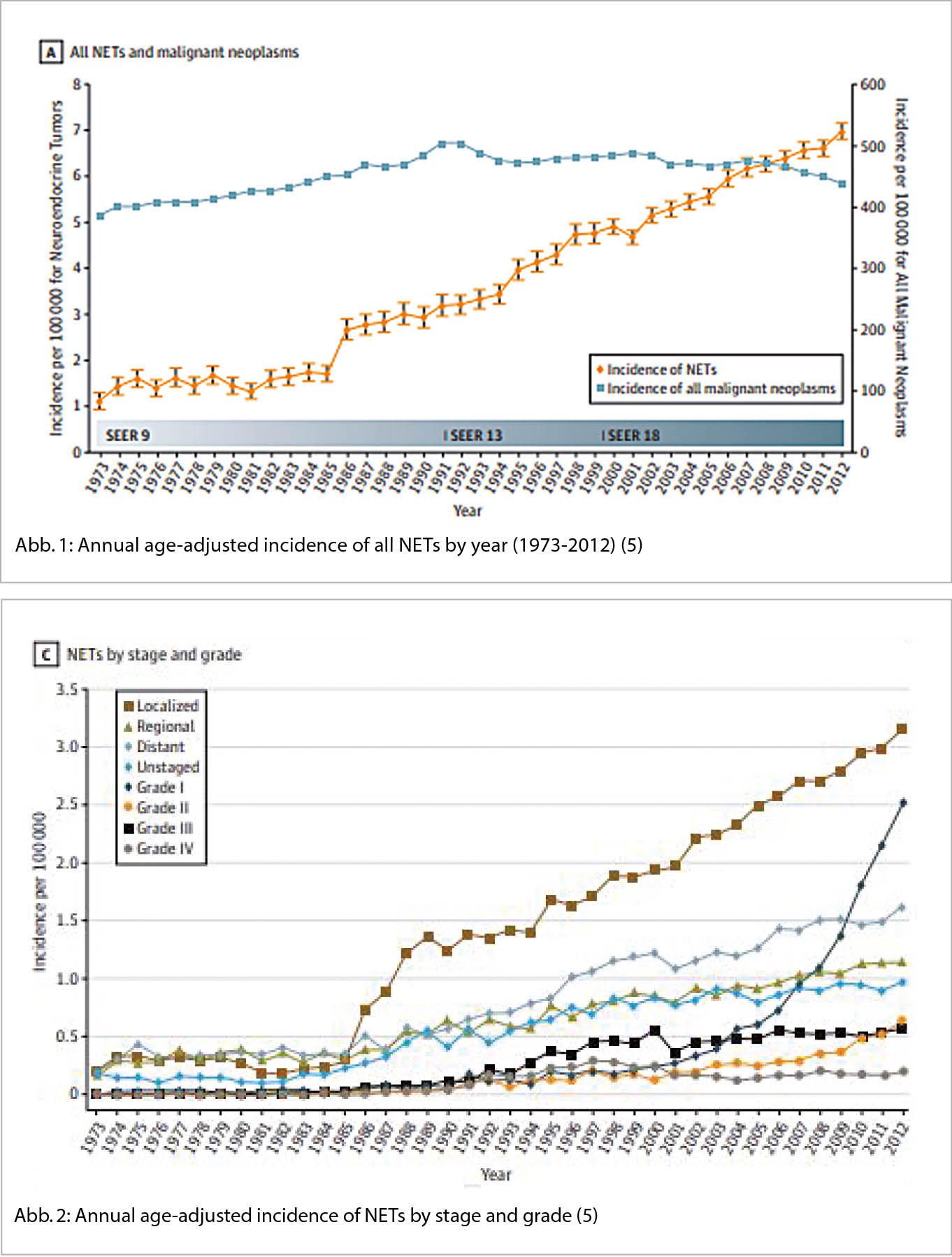
Die Inzidenz der NET hat in den letzten Jahrzehnten deutlich zugenommen (4). In den USA betrug die Inzidenz anfangs Siebzigerjahre noch 1.09 pro 100 000 Personen und stieg auf 6.98 pro 100’00 Personen im Jahre 2012. (Abb. 1) (4).
Ausserdem ist die Prävalenz der NET im gleichen Zeitraum von 0.006% 1993 auf 0.048% 2012 angestiegen und reflektiert somit nicht nur die steigende Inzidenz sondern auch das langsame Wachstumsverhalten. Kongruente Daten gibt es auch für Kanada und diverse europäische Länder (5).
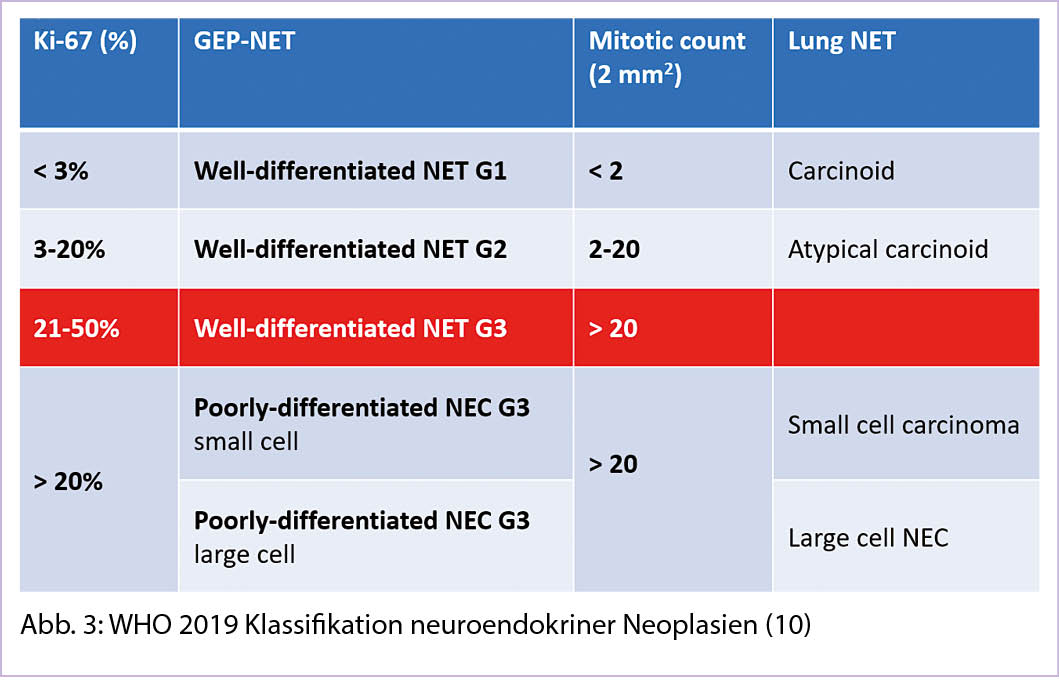
Auffällig ist die deutliche Inzidenzzunahme vor allem bei gut differenzierten NET im lokalisierten Stadium (Abbildung 2) (4). Fast zwei Drittel aller NET entstehen im Gastrointestinaltrakt und werden als gastroenteropankreatische NET (GEP-NEP) bezeichnet (6). Die Inzidenz und Prävalenz dieser wichtigen Untergruppe zeigt dieselbe epidemiologische Veränderung (7). Mit Blick auf die Primärlokalisation wiederholt sich das Muster, am wenigsten ausgeprägt ist die Zunahme bei den NET des Kolons (Annual percentage change APC 2.87), am auffälligsten im Rektum (APC 6.43) (7).
Obwohl bisher eine genaue Erklärung für diese epidemiologische Entwicklung fehlt, scheinen doch Trends in der modernen Medizin mit deutlicher Zunahme von eingesetzter Schnittbilddiagnostik (Ultraschall, CT, MRI) und verbesserte histologische Diagnostik eine wichtige Rolle bei der Zunahme der Inzidenz zu spielen. Die Zunahme der Prävalenz wird durch das verbesserte Überleben als Folge der sich entwickelnden Therapien erklärt.
Über die Hälfte aller NET befinden sich bei der Diagnosestellung in einem lokalisierten Stadium, 20% sind lokoregional fortgeschritten und gut 30% aller Patienten werden in einem metastasierten Stadium diagnostiziert (4). Allerdings variiert die Metastasierungsrate erheblich abhängig von der Primärlokalisation, während NET der Appendix fast nie metastasieren, sind Dünndarm-NET in 40-50% aller Neudiagnosen in einem Stadium M1 (8).
Klassifikation und Staging
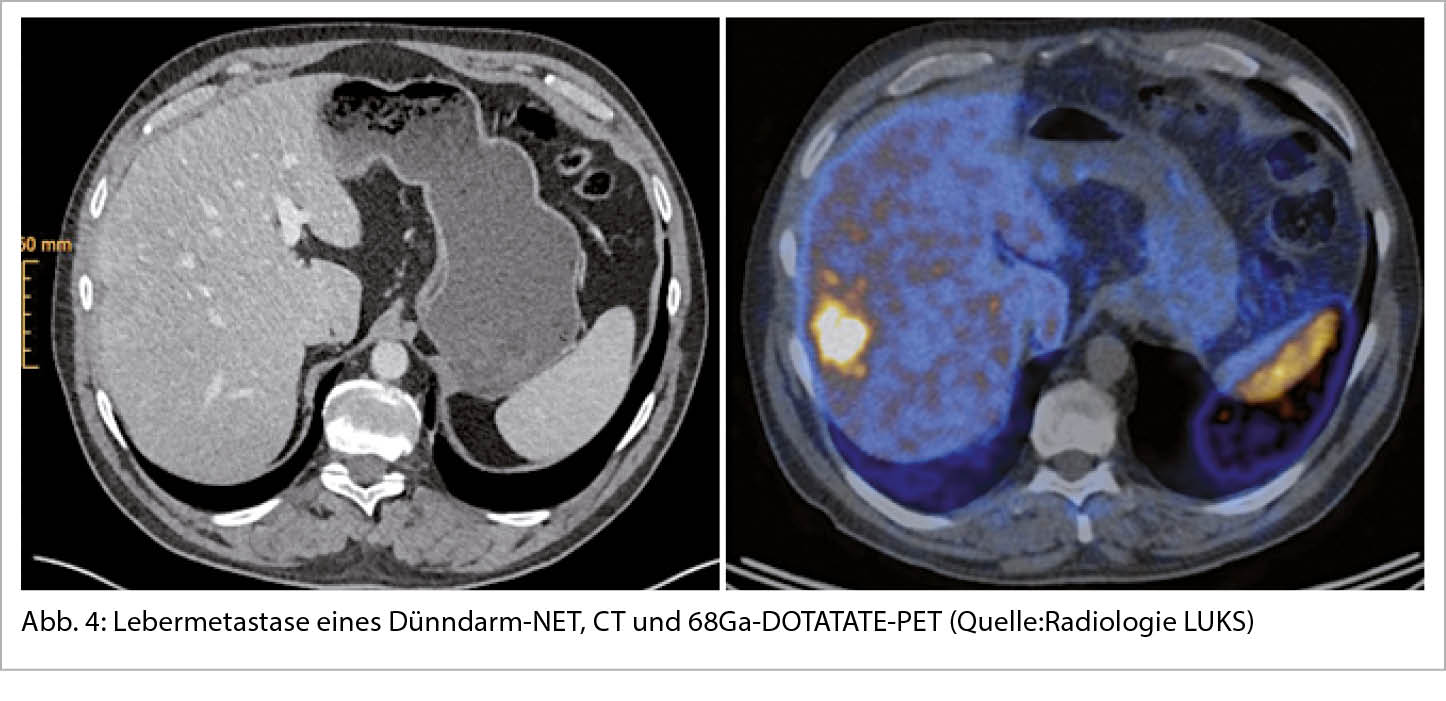
Seit der ersten Beschreibung eines neuroendokrinen Tumor des Dünndarms durch Langhans T. 1867 (9) hat sich die Klassifikation mehrmals gewandelt. Gerade 2019 wurde die neuste Version der WHO Klassifikation eingeführt, ein Ausdruck der Bemühungen, die Klassifikation für jede anatomische Lokalisation zu vereinheitlichen (10) (Abbildung 3). Die neuroendokrinen Neoplasien NEN werden unterteilt in die gut differenzierten neuroendokrinen Tumoren NET und die schlecht differenzierten neuroendokrinen Karzinome NEC. Dabei werden die NET mithilfe von Ki67 und mitotischer Aktivität in 3 Kategorien, G1-G3, unterteilt. Bei einer niedrigen Proliferation (Ki67<3%) spricht man von einem G1 Tumor, bei einem Ki67 von 3-20% von einem G2 und ab 21-50% von eine G3 Tumor. Eine Besonderheit in der Nomenklatur behalten die NET der Lunge, die G1 Tumoren bezeichnet man auch als typisches Karzinoid, ab G2 spricht man von einem atypischen Karzinoid. Bei den NEC unterscheidet man die klein- von den grosszelligen Histologien.
Die Unterscheidung dieser 2 Gruppen, NET und NEC, widerspiegelt nicht nur das klinische Verhalten sondern auch den molekularen Hintergrund: Die gut differenzierten NET zeigen häufig Mutationen in MEN1, DAXX oder ATRX, die NEC hingegen in TP53, Rb1 und SMAD4 (11).
Die meisten NEN sind gut differenziert und treten sporadisch auf. GEP-NETS und selten NET des Thymus und der Lunge können aber auch bei verschiedenen angeborenen Syndromen mit Keimbahnmutationen auftreten, zum Beispiel bei der multiplen endokrinen Neoplasie Typ 1 oder 2 (MEN1, 2), der von Hippel-Lindau Erkrankung (VHL), der tuberösen Sklerose (TSC) und der Neurofibromatose Typ 1 (NF-1) (12). Für die Stadieneinteilung hat sich die von der ENETS (European Neuroendocrine Tumour Society) erarbeitete TNM Klassifikation durchgesetzt und wurde in die 8. Ausgabe des UICC/AJCC Staging System übernommen (13).
Diagnose und Work-up
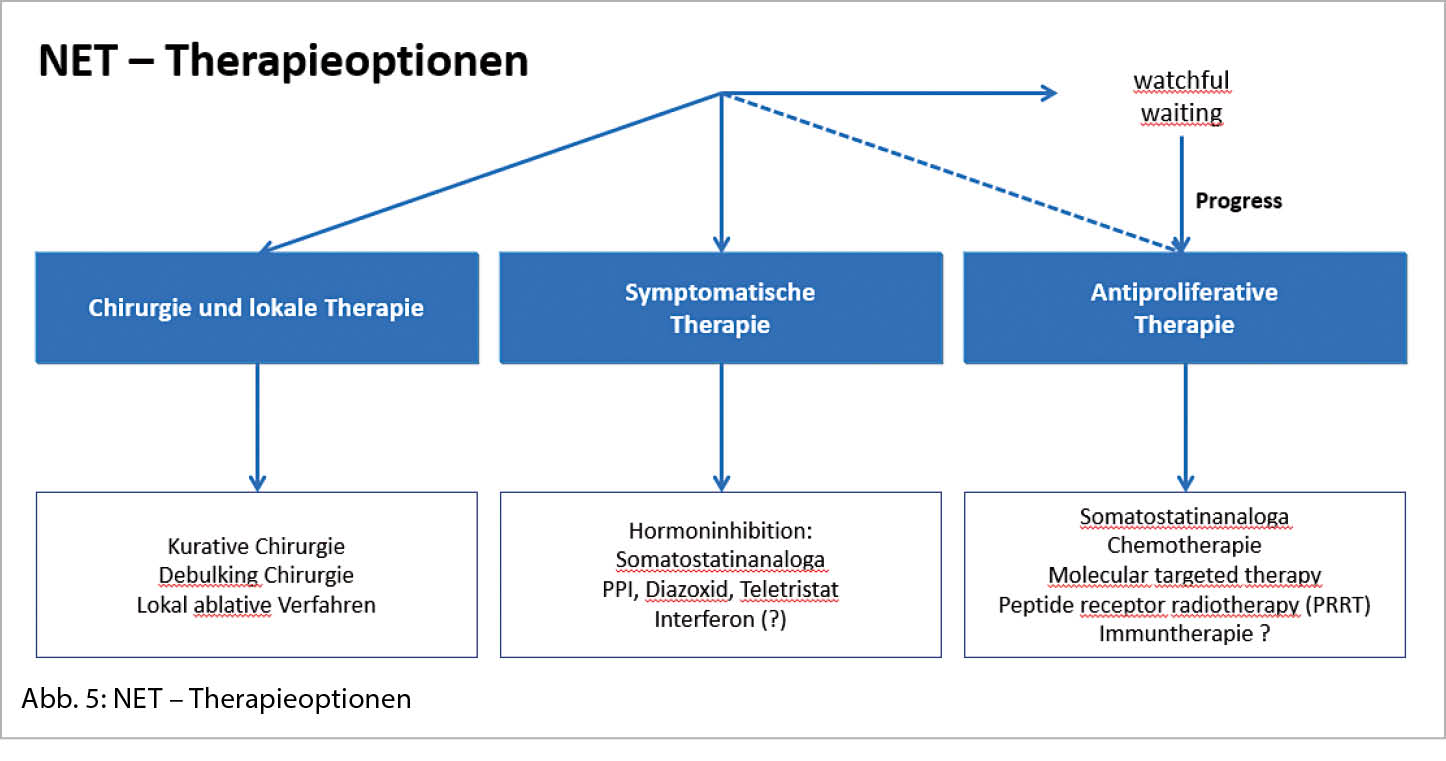
In der bildgebenden Diagnostik spielen die Schnittbildverfahren wie CT und MRI eine grundlegende Rolle sowohl für die Darstellung des Primarius als auch der Metastasierung. Mit der Computertomographie wird das Ausmass der Metastasierung, vor allem in der Leber oftmals unterschätzt, weshalb die MRI-Untersuchung der Leber mit einer deutlich höheren Sensitivität komplementär eingesetzt werden kann (14). Die Mehrheit der gut-differenzierten NET exprimiert Somatostatinrezeptoren (SSTR) an ihrer Zelloberfläche, vor allem den Somatostatinrezeptor-2 (SSTR2) (15). Mit der funktionellen Bildgebung, dem 68Ga-DOTATATE-PET gelingt nicht nur ein besseres Erfassen unbekannter Primärtumoren und Staging (auch ossärer und peritonealer Metastasen, Abbildung 4), sondern auch eine Darstellung des SSTR2, dem wichtigsten therapeutischen Target (16). Bei zunehmender Agressivität des Tumors (NET G3 oder gar NEC) nimmt die Somatostatinrezeptorexpression ab und die FDG-Affinität zu, sodass das FDG-PET als Ergänzung oder anstelle des DOTATATE-PET eingesetzt werden soll. Es wird empfohlen, anatomische und funktionelle Bildgebung in der Primärdiagnostik zu kombinieren.
Wie eingangs erwähnt, produzieren alle NET irgendwelche Peptide oder Hormone. Man unterscheidet aber funktionelle von nicht-funktionellen Tumoren. Je nach Literatur sind zwischen 25-30% der NET funktionell aktiv (2). Die hormonell aktiven Tumoren produzieren biochemisch aktive Substanzen in genügender Menge, dass daraus eine typische klinische Symptomatik resultiert. Das Karzinoid (Produktion von Serotonin) ist mit nahezu 80% aller funktionellen Syndrome das mit Abstand häufigste Syndrom, deutlich seltener sind Insulinome, Gastrinome, Glucagonome oder VIPome (17). Die nicht-funktionellen Tumoren produzieren meist hormonell-inaktive Peptide wie Chromogranin A oder Pankreatisches Polypeptid (PP). Diese können als Tumormarker verwendet werden, haben aber eine limitierte Sensitivität und Spezifität und werden durch verschiedene Medikamente (insbesondere PPIs) und Leber- und Nierenfunktionsstörungen beeinflusst (18). Die Einführung molekularer Biomarker könnte die eher unspezifischen Marker ablösen, zum Beispiel der NETest wird zur Zeit in prospektiven Studien auf seinen diagnostischen und prognostischen Wert hin evaluiert (19). Ein generelles Screening für Hormone wird bei asymptomatischen Patienten nicht empfohlen.
Therapie Übersicht
Um optimale Therapieentscheidungen fällen zu können, müssen die beschriebenen diagnostischen Schritte uns folgende Fragen beantworten: Handelt es sich um einen funktionalen oder nicht-funktionalen Tumor, ein lokalisiertes oder metastasiertes Stadium, einen low Grade oder high Grade Tumor mit stabilem Verhalten oder rascher Progredienz, besteht eine Somatostatinrezeptor Expression und woher stammt die Krankheit?
Zur Verfügung stehen uns chirurgische und lokale Therapieoptionen, symptomatische und antiproliferative Therapien (Abb. 5).
Chirurgie und lokale Therapien
Die radikale Resektion ist die einzige kurative Therapie bei lokalisierten oder lokoregional fortgeschrittenen NET mit guter Differenzierung und geringer Proliferation (G1-2). Symptome bei funktionellen Tumoren sollten vor der Operation kontrolliert werden (zum Beispiel Hypoglykämiekontrolle vor Resektion eines Insulinoms).
Pankreatische NET sollten wenn immer möglich reserziert werden (Standard-Pankreatektomie mit regionaler Lymphadenektomie), wenn sie eines der folgenden Kriterien erfüllen: Tumorgrösse > T1 (> 2cm), Zeichen lokaler Invasion (Dilatation Ductus choledochus, Ikterus), nodaler Befall, funktioneller Tumor (20). Für Tumoren die < 1cm messen und diese Kriterien nicht erfüllen, ist ein nicht-chirurgisches Vorgehen mit Beobachtung empfohlen (21). Widersprüchliche Daten existieren für Tumoren zwischen 1 und 2 cm Grösse (22, 23). Ein konservatives Vorgehen wird für ältere Patienten mit Komorbiditäten empfohlen und sollte bei jüngeren Patienten im Sinne eines «shared decision making» zumindest diskutiert werden (20).
Das empfohlene chirurgische Vorgehen bei NET des Dünndarms ist eine segmentale Resektion (oder ileozökale Resektion ilealer NET) mit Entfernung der regionalen Lymphknoten (24). Je nach Lokalisation der Lymphknoten in Bezug zur mesenterialen Gefässachse, kann dies sehr einfach bis technisch unmöglich sein (25). Intraoperativ sollte ein peritonealer Befall systematisch gesucht werden, ebenso ein möglicher multifokaler Dünndarmbefall.
Auch in ausgewählten metastasierten Situationen, mit resektablem Primärtumor und Metastasen, wird eine radikale Resektion von den ESMO-Guidelines empfohlen (20). Bei Patienten, die trotz systemischer Therapie eine Progression zeigen und vor allem unter einer ausgeprägten Lebermetastasierung leiden, können lokal-ablative Verfahren oder Metastasektomie im Sinne eines Debulking eingesetzt werden. Dies insbesondere auch bei funktionellen Tumoren mit ungenügender Symptomkontrolle (26).
Symptomatische Therapie
Das Karzinoid mit seiner klassischen Symptomatik (Flush, Diarrhoe, Bauchkrämpfen, Bronchospasmen und der Langzeitkomplikation, dem Hedinger Syndrom) ist der häufigste funktionelle NET (27). Im symptomatischen Management spielt die Instruktion der Patienten, Trigger zu vermeiden, eine wichtige Rolle. Dabei sollten amin-reiche Nahrungsmittel wie Schokolade, Bananen, Avokados, Kiwis, Nüsse und auch Alkohol gemieden werden. Das Fundament der Behandlung ist der Einsatz der Somatostatin-Analoga (SSA) wie Octreotid oder Lanreotid. Diese hemmen die Sekretion der verursachenden Hormone und führen bei 70-80% der Patienten zu einer Verbesserung der klinischen Symptomatik. Bei ungenügendem Effekt mit der Zieldosis (Octreotide LAR 30mg im. alle 4 Wochen oder Lanreotide LAR 120 mg sc. alle 4 Wochen) kann entweder die Dosis weiter gesteigert oder das Intervall verkürzt werden. Nach einer längeren Zeit mit guter klinischer Kontrolle kommt es bei vielen Patienten zu einer symptomatischen Progression, die den Einsatz von Telotristat ethyl (ein oraler Serotoninsynthese-Inhibitor, 28, 29) notwendig macht. Daten aus der RADIANT-2 Studie zeigen auch für Everolimus einen symptomatischen Effekt (30) und bei ausgeprägter Lebermetastasierung kann auch eine Zytoreduktion chirurgisch oder lokal-ablativ erwogen werden (31). Alternativ zeigen die Radionuclidtherapien (peptide receptor radionuclide therapy PRRT) eine Verbesserung der Symptomkontrolle (32).
Bei den Insulinomen steht die Vermeidung schwerer Hypoglykämien im Vordergrund. Instruktion zur Einnahme häufiger Mahlzeiten, eventuell der Einsatz von Dexamethason, Diazoxid und Everolimus müssen in Betracht gezogen werden. Wenn nicht kurativ resektabel, kann auch hier ein Tumordebulking diskutiert werden.
Bei einem VIPom mit massiver wässriger Diarrhoe, spielt erneut der Einsatz von SSA neben Flüssigkeits- und Elektrolytersatz die entscheidende Rolle. Bei den Gastrinomen ist der hochdosierte Einsatz von Protonenpumpen-Inhibitoren die Therapiegrundlage.
Antiproliferative Therapie
Nicht alle NET im metastasierten Stadium benötigen eine Therapie. Asymptomatische Patienten mit hochdifferenzierten Tumoren, niedriger Tumorlast und fehlender Tumorprogredienz können unter Umständen über lange Zeiträume nur beobachtet werden (watch and wait, 20).
Somatostatinanaloga SSA
Die SSA Octreotide und Lanreotide zeigen sowohl eine gute Langzeitverträglichkeit als auch symptomatische Wirkung bei funktionellen Tumoren. Die Nebenwirkungen der SSA-Therapie sind in aller Regel sehr milde. Nausea, abdominale Krämpfe, Durchfall und Steatorrhoe, Hyperglykämie und Cholelithiasis sind als die wichtigsten Nebenwirkungen zu nennen. In 2 randomisierten Studien, der PROMID Studie (33) und der CLARINET Studie (34) konnte sowohl für Octreotide als auch Lanreotide ein antiproliferativer Effekt gegenüber Placebo nachgewiesen werden. In der PROMID Studie mit 85 Patientin (midgut NET, gut differenziert, niedrige Tumorlast) verbesserte sich das progressionsfreie Überlebens PFS von 6.0 auf 14.3 Monate. In der CLARINET Studie mit 204 Patienten (nicht-funktionelle pankreatische und intestinale NET, G1 und G2 differenziert) zeigte sich eine PFS-Verbesserung von 18 auf 32.8 Monate. In beiden Studien konnte auch im Langzeitverlauf bei einem Cross-over von nahezu 90% keine Verbesserung des Gesamtüberlebens OS nachgewiesen werden. Gemäss der aktuellen ESMO-Guideline (20) sind Somatostatinanaloga die meist empfohlene Erstlinientherapie sowohl bei funktionellen wie nicht-funktionellen gastro-entero-pankreatischen NET GEP-NET) mit niedriger Proliferation.
Chemotherapie
Die systemische Chemotherapie ist bei fortgeschrittenen pankreatischen NET (PanNET) und G3-Tumoren (NET oder NEC) aller Lokalisationen indiziert (20). Bei den neuroendokrinen Karzinomen (NEC G3) ist die platin-basierte Chemotherapie weiterhin die bevorzugte Erstlinientherapie, in weiteren Linien kommen bei den gastro-entero-pankreatischen NEC Kombinationen wie FOLFOX, FOLFIRI und Temozolomid mit Capecitabine zum Einsatz (35). Bei den PanNET wurde schon in den 80iger Jahren die alkylierende Substanz Streptozocin in Kombinationen mit Fluorouracil oder Doxorubicin eingesetzt (36). Aufgrund der Toxizität wird diese Therapie aber heute selten verwendet. In retrospektiven Analysen zeigte sich eine vielversprechende Aktivität Temozolomid-basierter Therapien, insbesondere in der Kombination mit Capecitabine CAPTEM (37). In einer prospektiven Phase II Studie mit 145 Patienten mit progredienten PanNET wurde die Kombination CAPTEM mit Temozolomid als Monotherapie verglichen (38). Dabei verbesserte sich im Kombinationsarm das PFS auf 22.7 Monate (14.4 Monate im Monotherapiearm, HR 0.58, p = 0.023). Der CAPTEM Arm zeigte eine Ansprechrate von 33%., das mittlere Gesamtüberleben war zum Zeitpunkt der präsentierten Analyse nicht erreicht (OS mit TEM 38 Monate, HR 0.41, p = 0.012).
Everolimus
Der orale mTOR-Inhibitor Everolimus (10mg/d) wurde in den diversen RADIANT-Studien bei bronchialen und gastro-entero-pankreatischen NET untersucht. In der randomisierten RADIANT-3 Studie wurden 410 Patienten mit progredienten, metastasierten PanNET mit Everolimus oder Placebo behandelt und das PFS wurde von 4.6 auf 11.0 Monate verlängert (HR 0.35, p<0.001) (39). Das Gesamtüberleben wurde nicht signifikant verbessert (44.0 Monate versus 37.7 Monate, HR 0.94, p=0.30), vielleicht durch einen Cross-over Effekt. In der RADIANT-4 Studie wurden 302 Patienten mit nicht-funktionellen gastrointestinalen oder bronchialen NET eingeschlossen. Erneut zeigte sich eine Verbesserung des PFS (3.9 versus 11.0 Monate, HR 0.48, p<0.001) ohne einen Überlebensvorteil (40). Dem gegenüber stehen die bekannten Nebenwirkungen mit Stomatitiden, Hautveränderungen, Diarrhoe. Die Hyperglykämie als Nebenwirkung ist andererseits bei der Behandlung der Insulinome sehr erwünscht .
Tyrosinkinase-Inhibitoren TKI
Die erste Studie mit dem TKI Sunitinib wurde schon 2011 publiziert (41). Dabei wurden 171 Patienten mit PanNET zu Sunitinib (37.5mg/d) oder Placebo randomisiert und es konnte eine Verbesserung des PFS (5.5 versus 11,4 Monate, HR 0.42, p<0.001) gezeigt werden. Die objektive Ansprechrate lag unter 10%, ein Gesamtüberlebensvorteil fand sich nicht.
In 2 randomisierten Phase III Studien wurde der TKI Surufatinib untersucht. In der SANET-p Studie wurden 172 Patienten mit PanNET untersucht und das PFS von 3.7 Monaten mit Placebo signifikant auf 10.9 Monate verbessert (42). Die objektive Ansprechrate betrug interessante 19%. In der SANET-ep Studie wurden 289 Patienten mit extrapankreatischen NET eingeschlossen, erneut mit Verbesserung des PFS (3.8 versus 9.2 Monate, p<0.001) aber nur einer Ansprechrate von 10% (43). Der TKI Axitinid wurde in der Phase II/III Studie AXINET bei Patienten mit extra-pankreatischen NET untersucht und zeigt eine hohe Ansprechrate von 17.5%, jedoch keine signifikante Verbesserung des PFS (12.3 versus 17.2 Monate, p=0.169) (44). Interessante Phase II Daten gibt es für Lenvatinib (45) und zur Zeit wird Cabozantinib in der Phase III CABINET Studie untersucht.
Peptide Receptor Radionuclide Therapy PRRT
Seit nahezu 30 Jahren haben wir in der Schweiz Erfahrung mit der PRRT in der Form Yttrium90 oder Lutetium177 gebunden an DOTATATE oder DOTATOC bei der Behandlung von neuroendokrinen Tumoren (46). Für die Therapie qualifizieren Patienten, deren Tumoren eine starke und homogene Expression des Somatostatinrezeptor-2 (SSTR2) zeigen. Dies wird heute üblicherweise mit einem Dotatate-PET gemessen. Daten für die Behandlung von pankreatischen NET kommen vor allem aus dem ERASMUS Projekt und zeigen eine Ansprechrate von 16% und eine mediane Ansprechsdauer von 35 Monaten (47). Für die nicht-pankreatischen Midgut NET wurde 2017 die randomisierte Phase III Studie NETTER-1 publiziert (48). Dabei wurden 229 Patienten zu 177Lu-DOTATATE oder high-dose Octreotide randomisiert. Das progressionsfreie Überleben wurde von 8.5 auf 28.4 Monate verbessert und eine objektive Ansprechrate von 18% erreicht. In der finalen Analyse zeigte sich gegenüber der Originalpublikation kein signifikanter Überlebensvorteil mehr (median OS 36.3 versus 48.0 Monate, HR 0.84, p=0.03), dies möglicherweise durch einen Crossover von 36%. Zu beachtende Nebenwirkungen sind Hämatotoxität, Nephrotoxizität (deutlich geringer mit Lutetium als mit Yttrium) und selten die Entwicklung eines myelodysplastischen Syndroms oder einer Leukämie (47). Mindestens 4 Wochen vor einer PRRT sollten Octreotide LAR oder Lanreotide gestoppt werden und im Bedarfsfall (funktioneller Tumor) auf die kurzwirksame, subkutane Therapie umgestellt werden.
Neue Therapien und Studien
Wie bei jeder Tumorentität wird auch bei den NEN die Immuntherapie mit Checkpoint-Inhibitoren untersucht. Die bisherige Datenlage für gut differenzierte NET ist sowohl für die Monotherapien mit Pembrolizumab (KN 158) oder Spartalizumab (E2201) oder Kombinationen (DUNE mit Durvalumab/Tremelimumab oder DART mit Ipi/Nivo) wenig überzeugend. Es zeigen sich Ansprechraten von 0 bis 6.9%. Allerdings handelt es sich bisher um sehr kleine Studien mit heterogenen Populationen. Interessanter sind die Daten bei den G3 Tumoren, sind aber widersprüchlich mit einer Ansprechrate von 44% mit Ipi/Nivo (DART) und 7.2% mit Durvalumab/Tremelimumab (DUNE). Bisher kann ein Einsatz ausserhalb von Studien nicht empfohlen werden. Mit Spannung werden vor allem Daten zur Sequenzierung (COMPEDE: Everolimus vs.PRRT 2nd line, SEQTOR: Everolimus vs.Chemotherapie, OCULORANDOM: PRRT vs.Sunitinib) erwartet. Von diversen Kombinationen (z.B. PRRT mit Chemotherapie, TKIs und Immunotherapie) werden weitere Therapieverbesserungen erhofft. Obwohl in den letzten 10 Jahren zunehmend auch randomisierte Studien bei NEN durchgeführt werden, würde eine Fokussierung auf genauer definierte Populationen anstelle von Basket-Studien die klinische Entwicklung neuer Therapieansätze fördern (49).
Therapiesequenzierung und Guidelines
Klare Behandlungsindikationen sind gegeben durch eine G3-Differenzierung (NET und NEC), eine Tumorprogression und symptomatische Situationen, sei es durch die Tumorlast oder die Hormonaktivität. In der Literatur findet sich kein klarer Konsens über die optimale Sequenzierung der beschriebenen Therapieoptionen. Eine gute Übersicht und Hilfe im klinischen Alltag bietet die 2020 publizierte ESMO-Guideline (20). Dabei wird abhängig von Primärtumor, SSTR-Expression, Ki67 und Wachstumsdynamik eine Empfehlung für die Wahl der Erst-, Zweit- und Drittlinientherapie gegeben.
Copyright bei Aerzteverlag medinfo AG
Co-Chefarzt/Leiter Onkologie Sursee
Luzerner Kantonsspital | Tumorzentrum
Spitalstrasse | 6000 Luzern 16
Spitalstrasse | 6210 Sursee
Der Autor hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
1. öberg, K. E. (2010). Gastrointestinal neuroendocrine tumors. Annals of Oncology, 21, vii72-vii80.
2. Cives, M., & Strosberg, J. R. (2018). Gastroenteropancreatic neuroendocrine tumors. CA: a cancer journal for clinicians, 68(6), 471-487.
3. Singh, S., Granberg, D., Wolin, E., Warner, R., Sissons, M., Kolarova, T., … & Leyden, J. (2017). Patient-reported burden of a neuroendocrine tumor (NET) diagnosis: results from the first global survey of patients with NETs. Journal of global oncology, 3(1), 43-53.
4. Dasari, A., Shen, C., Halperin, D., Zhao, B., Zhou, S., Xu, Y., … & Yao, J. C. (2017). Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA oncology, 3(10), 1335-1342.
5. Huguet, I., Grossman, A. B., & O’Toole, D. (2017). Changes in the epidemiology of neuroendocrine tumours. Neuroendocrinology, 104(2), 105-111.
6. Cives, M., & Strosberg, J. R. (2018). Gastroenteropancreatic neuroendocrine tumors. CA: a cancer journal for clinicians, 68(6), 471-487.
7. Xu, Z., Wang, L., Dai, S., Chen, M., Li, F., Sun, J., & Luo, F. (2021). Epidemiologic Trends of and Factors Associated With Overall Survival for Patients With Gastroenteropancreatic Neuroendocrine Tumors in the United States. JAMA network open, 4(9), e2124750-e2124750.
8. Frilling, A., Modlin, I. M., Kidd, M., Russell, C., Breitenstein, S., Salem, R., … & Schilsky, R. (2014). Recommendations for management of patients with neuroendocrine liver metastases. The lancet oncology, 15(1), e8-e21.
9. Langhans T. (1867). Ueber einen Drüsenpolyp im Ileum. Virchows Arch 38:559-560
10. WHO Classification of Tumours Editorial Board: Digestive System Tumours, WHO Classification of Tumours (ed 5). Lyon, France, International Agency for Research on Cancer, 2019
11. Tang, L. H., Basturk, O., Sue, J. J., & Klimstra, D. S. (2016). A practical approach to the classification of WHO grade 3 (G3) well differentiated neuroendocrine tumor (WD-NET) and poorly differentiated neuroendocrine carcinoma (PD-NEC) of the pancreas. The American journal of surgical pathology, 40(9), 1192.
12. Shah, M. H., Goldner, W. S., Halfdanarson, T. R., Bergsland, E., Berlin, J. D., Halperin, D., … & Zuccarino-Catania, G. (2018). NCCN guidelines insights: neuroendocrine and adrenal tumors, version 2.2018. Journal of the National Comprehensive Cancer Network, 16(6), 693-702.
13. Brierley, J. D., Gospodarowicz, M. K., & Wittekind, C. (Eds.). (2017). TNM classification of malignant tumours. John Wiley & Sons.
14. d’Assignies, G., Fina, P., Bruno, O., Vullierme, M. P., Tubach, F., Paradis, V., … & Vilgrain, V. (2013). High sensitivity of diffusion-weighted MR imaging for the detection of liver metastases from neuroendocrine tumors: comparison with T2-weighted and dynamic gadolinium-enhanced MR imaging. Radiology, 268(2), 390-399.
15. Reubi, J. C., Kvols, L. K., Waser, B., Nagorney, D. M., Heitz, P. U., Charboneau, J. W., … & Moertel, C. (1990). Detection of somatostatin receptors in surgical and percutaneous needle biopsy samples of carcinoids and islet cell carcinomas. Cancer research, 50(18), 5969-5977.
16. Sadowski, S. M., Neychev, V., Millo, C., Shih, J., Nilubol, N., Herscovitch, P., … & Kebebew, E. (2016). Prospective study of 68Ga-DOTATATE positron emission tomography/computed tomography for detecting gastro-entero-pancreatic neuroendocrine tumors and unknown primary sites. Journal of Clinical Oncology, 34(6), 588.
17. Falconi, M., Eriksson, B., Kaltsas, G., Bartsch, D. K., Capdevila, J., Caplin, M., … & Jensen, R. T. (2016). ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology, 103(2), 153-171.
18. Raines, D., Chester, M., Diebold, A. E., Mamikunian, P., Anthony, C. T., Mamikunian, G., & Woltering, E. A. (2012). A prospective evaluation of the effect of chronic proton pump inhibitor use on plasma biomarker levels in humans. Pancreas, 41(4), 508-511.
19. Modlin, I. M., Kidd, M., Bodei, L., Drozdov, I., & Aslanian, H. (2015). The clinical utility of a novel blood-based multi-transcriptome assay for the diagnosis of neuroendocrine tumors of the gastrointestinal tract. Official journal of the American College of Gastroenterology| ACG, 110(8), 1223-1232.
20. Pavel, M., Öberg, K., Falconi, M., Krenning, E. P., Sundin, A., Perren, A., & Berruti, A. (2020). Gastroenteropancreatic neuroendocrine neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology, 31(7), 844-860.
21. Kulke, M. H., Anthony, L. B., Bushnell, D. L., De Herder, W. W., Goldsmith, S. J., Klimstra, D. S., … & Jensen, R. T. (2010). NANETS treatment guidelines: well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas, 39(6), 735.
22. Lee, L. C., Grant, C. S., Salomao, D. R., Fletcher, J. G., Takahashi, N., Fidler, J. L., … & Huebner, M. (2012). Small, nonfunctioning, asymptomatic pancreatic neuroendocrine tumors (PNETs): role for nonoperative management. Surgery, 152(6), 965-974.
23. Sun, Y., Wang, Y., Li, R., Kang, G., Zhang, M., Chen, X., … & Yu, Q. (2021). Surgical resection of primary tumor is associated with prolonged survival in low-grade pancreatic neuroendocrine tumors. Clinics and research in hepatology and gastroenterology, 45(1), 101432.
24. Scott, A. T., & Howe, J. R. (2018). Management of small bowel neuroendocrine tumors. Journal of oncology practice, 14(8), 471-482.
25. Lardière-Deguelte, S., de Mestier, L., Appéré, F., Vullierme, M. P., Zappa, M., Hoeffel, C., … & Kianmanesh, R. (2016). Toward a preoperative classification of lymph node metastases in patients with small intestinal neuroendocrine tumors in the era of intestinal-sparing surgery. Neuroendocrinology, 103(5), 552-559.
26. Liu, D. M., Kennedy, A., Turner, D., Rose, S. C., Kee, S. T., Whiting, S., … & Salem, R. (2009). Minimally invasive techniques in management of hepatic neuroendocrine metastatic disease. American journal of clinical oncology, 32(2), 200-215.
27. Rubin de Celis Ferrari, A. C., Glasberg, J., & Riechelmann, R. P. (2018). Carcinoid syndrome: update on the pathophysiology and treatment. Clinics, 73.
28. Kulke, M. H., Hörsch, D., Caplin, M. E., Anthony, L. B., Bergsland, E., Öberg, K., … & Pavel, M. (2017). Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome. Journal of clinical oncology, 35(1), 14.
29. Pavel, M., Gross, D. J., Benavent, M., Perros, P., Srirajaskanthan, R., Warner, R. R., … & Garcia-Carbonero, R. (2018). Telotristat ethyl in carcinoid syndrome: safety and efficacy in the TELECAST phase 3 trial. Endocrine-related cancer, 25(3), 309-322.
30. Bainbridge, H. E., Larbi, E., & Middleton, G. (2015). Symptomatic control of neuroendocrine tumours with everolimus. Hormones and Cancer, 6(5), 254-259.
31. Strosberg, J. R., Choi, J., Cantor, A. B., & Kvols, L. K. (2006). Selective hepatic artery embolization for treatment of patients with metastatic carcinoid and pancreatic endocrine tumors. Cancer Control, 13(1), 72-78.
32. Pathirana, A. A., Vinjamuri, S., Byrne, C., Ghaneh, P., Vora, J., & Poston, G. J. (2001). 131I-MIBG radionuclide therapy is safe and cost-effective in the control of symptoms of the carcinoid syndrome. European Journal of Surgical Oncology (EJSO), 27(4), 404-408.
33. Rinke, A., Wittenberg, M., Schade-Brittinger, C., Aminossadati, B., Ronicke, E., Gress, T. M., … & PROMID Study Group. (2017). Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors (PROMID): results of long-term survival. Neuroendocrinology, 104(1), 26-32.
34. Caplin, M. E., Pavel, M., Ćwikła, J. B., Phan, A. T., Raderer, M., Sedláčková, E., … & Ruszniewski, P. (2014). Lanreotide in metastatic enteropancreatic neuroendocrine tumors. New England Journal of Medicine, 371(3), 224-233.
35. McNamara, M. G., Frizziero, M., Jacobs, T., Lamarca, A., Hubner, R. A., Valle, J. W., & Amir, E. (2020). Second-line treatment in patients with advanced extra-pulmonary poorly differentiated neuroendocrine carcinoma: a systematic review and meta-analysis. Therapeutic Advances in Medical Oncology, 12, 1758835920915299.
36. Lamarca, A., Elliott, E., Barriuso, J., Backen, A., McNamara, M. G., Hubner, R., & Valle, J. W. (2016). Chemotherapy for advanced non-pancreatic well-differentiated neuroendocrine tumours of the gastrointestinal tract, a systematic review and meta-analysis: A lost cause?. Cancer treatment reviews, 44, 26-41.
37. Cives, M., Ghayouri, M., Morse, B., Brelsford, M., Black, M., Rizzo, A., … & Strosberg, J. (2016). Analysis of potential response predictors to capecitabine/temozolomide in metastatic pancreatic neuroendocrine tumors. Endocr Relat Cancer, 23(9), 759-767.
38. Kunz, P. L., Catalano, P. J., Nimeiri, H., Fisher, G. A., Longacre, T. A., Suarez, C. J., … & Benson, A. B. (2018). A randomized study of temozolomide or temozolomide and capecitabine in patients with advanced pancreatic neuroendocrine tumors: A trial of the ECOG-ACRIN Cancer Research Group (E2211).
39. Yao, J. C., Shah, M. H., Ito, T., Bohas, C. L., Wolin, E. M., Van Cutsem, E., … & Öberg, K. (2011). Everolimus for advanced pancreatic neuroendocrine tumors. New England Journal of Medicine, 364(6), 514-523.
40. Yao, J. C., Fazio, N., Singh, S., Buzzoni, R., Carnaghi, C., Wolin, E., … & Pavel, M. E. (2016). Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. The Lancet, 387(10022), 968-977.
41. Raymond, E., Dahan, L., Raoul, J. L., Bang, Y. J., Borbath, I., Lombard-Bohas, C., … & Ruszniewski, P. (2011). Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. New England Journal of Medicine, 364(6), 501-513.
42. Xu, J., Shen, L., Bai, C., Wang, W., Li, J., Yu, X., … & Su, W. (2020). Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): a randomised, double-blind, placebo-controlled, phase 3 study. The Lancet Oncology, 21(11), 1489-1499.
43. Xu, J., Shen, L., Zhou, Z., Li, J., Bai, C., Chi, Y., … & Su, W. (2020). Surufatinib in advanced extrapancreatic neuroendocrine tumours (SANET-ep): a randomised, double-blind, placebo-controlled, phase 3 study. The Lancet Oncology, 21(11), 1500-1512.
44. Garcia-Carbonero, R., Benavent, M., Jiménez Fonseca, P., Castellano, D., Alonso, T., Teule, A., … & Capdevila, J. (2021). A phase II/III randomized double-blind study of octreotide acetate LAR with axitinib versus octreotide acetate LAR with placebo in patients with advanced G1-G2 NETs of non-pancreatic origin (AXINET trial-GETNE-1107).
45. Capdevila, J., Fazio, N., Lopez Lopez, C., Teule, A., Valle, J. W., Tafuto, S., … & Ibrahim, T. (2019). Final results of the TALENT trial (GETNE1509): a prospective multicohort phase II study of lenvatinib in patients (pts) with G1/G2 advanced pancreatic (panNETs) and gastrointestinal (giNETs) neuroendocrine tumors (NETs).
46. Imhof, A., Brunner, P., Marincek, N., Briel, M., Schindler, C., Rasch, H., … & Walter, M. A. (2011). Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. Journal of clinical oncology, 29(17), 2416-2423.
47. Brabander, T., Van der Zwan, W. A., Teunissen, J. J., Kam, B. L., Feelders, R. A., de Herder, W. W., … & Kwekkeboom, D. J. (2017). Long-term efficacy, survival, and safety of [177Lu-DOTA0, Tyr3] octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine tumors. Clinical Cancer Research, 23(16), 4617-4624.
48. Strosberg, J., El-Haddad, G., Wolin, E., Hendifar, A., Yao, J., Chasen, B., … & Krenning, E. (2017). Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. New England Journal of Medicine, 376(2), 125-135.
49. Das, S., Du, L., Lee, C. L., Arhin, N. D., Chan, J. A., Kohn, E. C., … & Dasari, A. (2021). Comparison of Design, Eligibility, and Outcomes of Neuroendocrine Neoplasm Trials Initiated From 2000 to 2009 vs 2010 to 2020. JAMA network open, 4(10), e2131744-e2131744.

info@onco-suisse
- Vol. 12
- Ausgabe 2
- März 2022