Wir präsentieren einen Fall einer eosinophilen Granulomatose mit Polyangiitis (EGPA) und geben einen Überblick über eosinophile Lungenerkrankungen. Dabei beleuchten wir die diagnostischen und therapeutischen Herausforderungen dieser Krankheitsgruppe. Zudem diskutieren wir die Klassifizierung des Schweregrads der EGPA und bieten Einblicke in aktuelle Behandlungskonzepte.
Schlüsselwörter: Eosinophile Lungenerkrankungen, eosinophile Granulomatose mit Polyangiitis, Hypereosinophilie, Eosinophile
Anamnese und Befunde
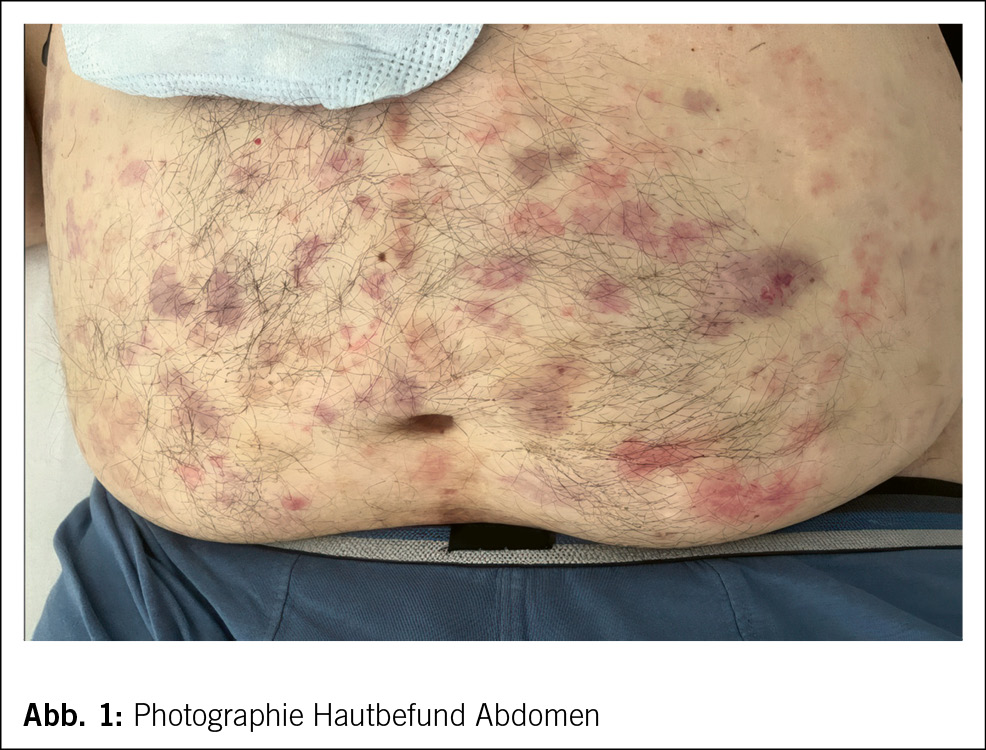
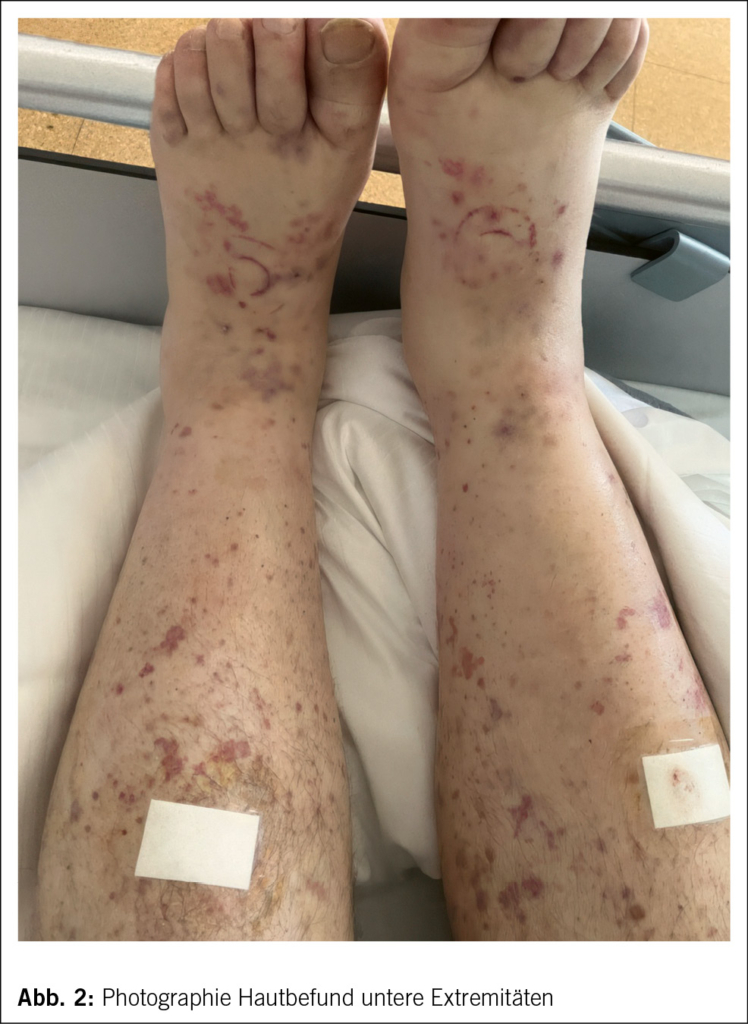
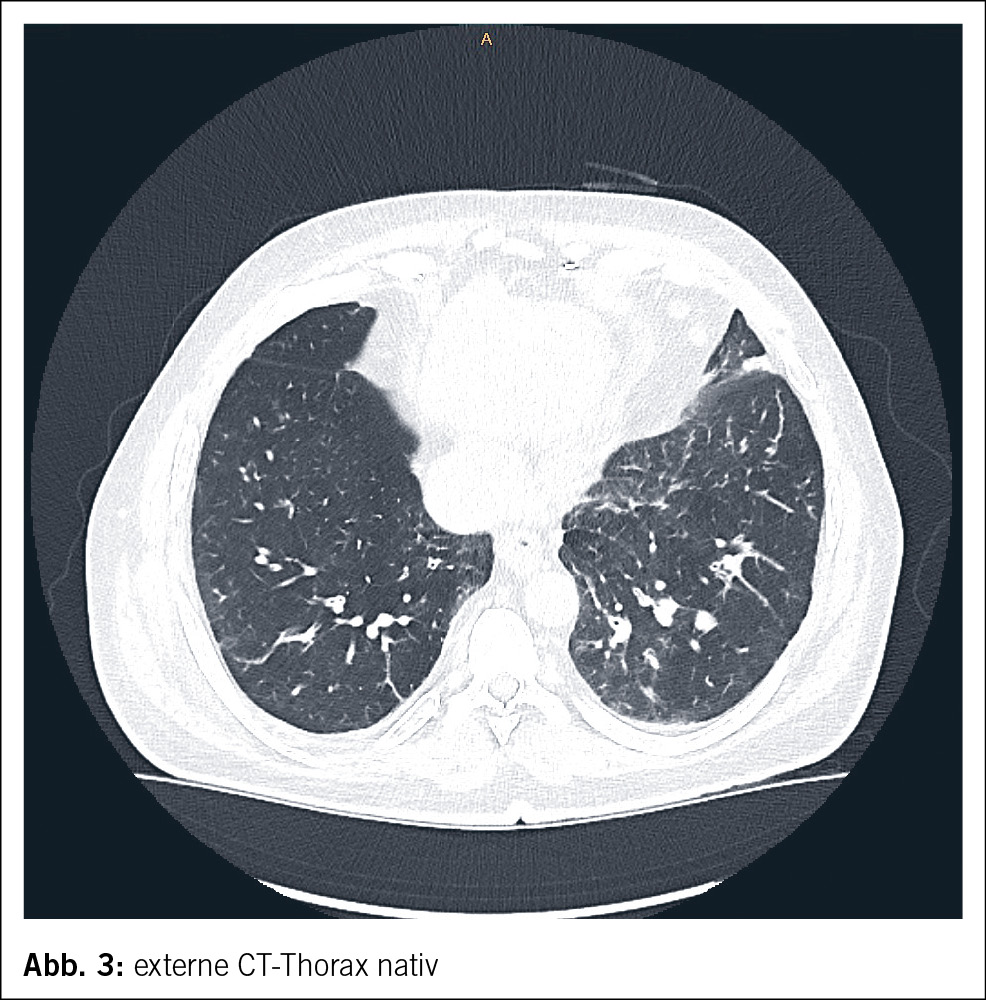
Ein 61-jähriger Mann wurde durch den Hausarzt bei Verdacht auf eine ambulant erworbene Pneumonie in ein wohnortnahes peripheres Spital zugewiesen. Neben purulentem Auswurf imponierten bei dortiger Aufnahme eine disseminierte leicht palpable Purpura (Abb. 1 und Abb. 2) mit enoraler Aussparung, ausgeprägte bilaterale Unterschenkelödeme sowie eine Bluteosinophilie von 3.1 G/l (0.0–0.7 G/l). In der CT-Thorax wurde eine basale Bronchiolitis mit weiteren bronchiolitischen Veränderungen im rechten Oberlappen ohne Konsolidationen oder Pleuraergüssen sowie Groundglass-Opazitäten in beiden Unterlappen nachgewiesen (Abb. 3). Eine antiinfektive Theapie mit Amoxicillin/Clavulansäure wurde initiiert, welche im Verlauf auf Ceftriaxon und Clarithromycin eskaliert wurde. Ausserdem veranlasste man eine Stanzbiopsie der Purpura im Bereich der streckseitigen Unterschenkel. Eine geplante diagnostische Bronchoskopie musste bei periinterventioneller hämodynamischer Instabilität und Sättigungsabfällen abgebrochen werden. Daraufhin erfolgte die Verlegung ins Universitätsspital Zürich zur weiteren pneumologischen Abklärung.
Kurze Anamnese mit Betonung des jetzigen Leidens
Bei Eintritt im Universitätsspital Zürich präsentierte sich ein 61-jähriger Patient in vermindertem, initial afebrilem Allgemeinzustand, kardiorespiratorisch stabil mit ausgeprägten bilateralen Unterschenkelödemen sowie leicht palpabler Purpura an Körperstamm und den unteren Extremitäten. Auskultatorisch imponierte ein 2/6 Systolikum mit punctum maximum über der Aortenklappe. Die Gelenke waren allseits schmerzfrei beweglich ohne palpable Synovitiden. Der Neurostatus war unauffällig. Im weiteren stationären Verlauf kam es zu täglichen Fieberepisoden bis max. 39.2 °C.
Der Patient berichtete, an einem langjährigen eosinophilen Asthma und einer Rhinosinusitis mit chronisch behinderter Nasenatmung zu leiden. An weiterer Vorerkrankung war eine valvuläre und koronare Dreigefässerkrankung mit Status nach biologischem Aortenklappenersatz und aortokoronarer Bypassoperation im Jahr 2015 bekannt.
In unserem Eintrittslabor sahen wir erneut eine Bluteosinophilie von 2.94 G/l, welche mikroskopisch bestätigt wurde. Im mikroskopischen Differenzialblutbild zeigten sich keine weiteren Auffälligkeiten, insbesondere kein Nachweis von Blasten. Es imponierte eine humorale Entzündungsaktivität mit einem C-reaktivem Protein von 96 mg/l (< 5mg/l), das Procalcitonin lag bei 0.13 µg/l (< 0.1 µg/l). Das Gesamt-IgE war mit > 2000 ku/l (< 100 ku/l) deutlich erhöht. Die ANCA-Serologie war negativ. In der Spirometrie fand sich eine fixierte obstruktive Ventilationsstörung mit einer FEV1/FVC 67 % bzw. einem Z-Score von –1.49 (> 75 %) ohne Diffusionsstörung. Als Leitsymptom berichtet der Patient über einen produktiven Husten sowie eine ausgeprägte und progrediente Belastungsdyspnoe.
Differenzialdiagnostische Überlegungen
Unser Patient präsentierte neben den beidseitigen pulmonalen Infiltraten eine Hypereosinophilie, die das differenzialdiagnostische Spektrum einer eosinophilen Lungenerkrankung eröffnet hat. Dieses beinhaltet eine heterogene Gruppe verschiedener Krankheitsentitäten. Als Gemeinsamkeit findet sich eine Infiltration von Eosinophilen innerhalb der Atemwege, Alveolen und/oder dem Lungeninterstitium (1). Des Weiteren wird häufig eine periphere Eosinophilie nachgewiesen.
Die definitive Diagnosestellung im Akutstadium sowie die Abgrenzung der verschiedenen Krankheitsentitäten untereinander stellen eine klinische Herausforderung dar. Nach der Diagnosesicherung einer eosinophilen Lungenerkrankung gilt es im Abklärungsalgorithmus darum, einen möglichen Auslöser und eine extrapulmonale Organbeteiligung zu evaluieren. Folgende Kriterien definieren eosinophile Erkrankungen mit Lungenbeteiligung (2): periphere Eosinophilie (absolute Eosinophilenzahl ≥ 500 Eosinophile/µl) und Opazitäten in der pulmonalen Bildgebung. Erhöhter Eosinophilenanteil in der bronchoalveolären Lavage (> 10 %) und/oder histologischer Nachweis von eosinophilen Entzündungszellen in den Atemwegen oder dem Lungengewebe.
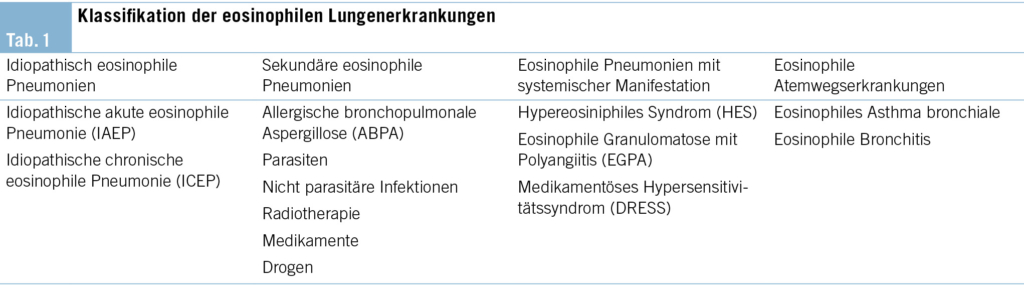
Bezüglich der Klassifikation sollte eine Unterteilung in akut und chronisch sowie eine pulmonale und systemische Manifestation erfolgen (Tab. 1) (3).
In unserem Fall kann das bekannte eosinophile Asthma bereits eine periphere Eosinophilie bedingen, ist allerdings auch als Begleiterkrankung bei anderen eosinophilen Lungenerkrankungen wie der chronischen eosinophilen Pneumonie, der eosinophilen Granulomatose mit Polyangiitis (EGPA) und des hypereosinophilen Syndroms (HES) beschrieben. Ein unkontrolliertes Asthma bronchiale in Verbindung mit steigenden eosinophilen Zellen und IgE-Titer muss differenzialdiagnostisch ebenfalls an eine allergische bronchopulmonale Aspergillose (ABPA) denken lassen. Eine Bestimmung der Aspergillus-spezifischen IgG- und IgE-Titer sollte hier erfolgen. Andere Auslöser einer peripheren Eosinophilie mit Lungenbeteiligung wie Infektionen (v.a. Parasiten, Medikamente oder Tumorerkrankungen) sollten anamnestisch eruiert und bei Verdacht weiter abgeklärt werden. Es gilt, weiterhin eine systemische Erkrankung mit extrapulmonaler Manifestation zu erfassen. Hierfür müssen eine breite Systemanamnese inklusive Beachtung etwaiger Vaskulitiszeichen, eine weitreichende körperliche Untersuchung inklusive Neurostatus, Labor -, Funktionsdiagnostik und Bildgebung erfolgen. Ein extrapulmonaler eosinophiler Organbefall schliesst eine chronische eosinophile Pneumonie aus und bedingt die Fokussierung auf die differenzialdiagnostische Evaluation einer EGPA und eines HES. Diese mitunter schwierige Differenzierung wird mit der Evaluation der Wahrscheinlichkeit eines primären (klonalen) HES begonnen. Hierfür bietet sich die Bestimmung von Tryptase und Vitamin B12, ein Hepatomegalie- Screening sowie ein mikroskopisches Blutbild mit Frage nach Blasten an (5). Auch eine steroid-refraktäre Eosinophilie sollte an ein HES denken lassen. Ist die Vortestwahrscheinlichkeit hoch, sollte im Rahmen einer Stufendiagnostik erst ein Screening auf die häufigste molekulargenetische Aberration, FIP1L1-PDGFRA-Rearrangement, mittels PCR oder FISH erfolgen, bevor Knochenmarkbiopsie und weitere zytogenetische oder molekulargenetische Untersuchungen folgen. Bei niedriger Vortestwahrscheinlichkeit oder klarem Surrogat einer Vaskulitis sollte eine EGPA abgeklärt werden. Es ist zu bedenken, dass es keine validierten einheitlichen Diagnostikkriterien für die EGPA gibt. Wechsler et al. etablierten im Rahmen des Mirra Trial Einschlusskriterien (6), die bei der Diagnose einer EGPA helfen können. Diese Kriterien umfassen Asthma und Bluteosinophilie (>1.0 G/l) sowie zwei der folgenden Faktoren:
– histologischer Nachweis einer eosinophilen Vaskulitis, perivaskuläre eosinophile Infiltrate, eosinophile, granulomatöse Entzündung
– Neuropathie
– sinunasaler Befall
– Kardiomyopathie
– Glomerulonephritis
– alveoläre Hämorrhagie
– palpable Purpura
– positive ANCA
Es ist zu bedenken, dass nur ca. 30–40 % der EGPA-Patienten positive ANCA (meist p-ANCA) aufweisen. Diese Patienten werden dem vaskulitischen Phänotyp des ANCA-Spektrums zugeordnet und präsentieren häufiger Nierenbeteiligung, Mononeuritis und Purpura.
Die ACR/EULAR-Kriterien erlauben zwar eine Differenzierung gegenüber anderen Vaskulitiden, aber keine de novo EGPA-Diagnose (7). Unser Patient zeigte mit der Purpura Zeichen einer kutanen Vaskulitismanifestation, die histologisch als solche bestätigt werden konnte.
Weitere Abklärungsschritte
Zur differenzialdiagnostischen Abklärung wurden Blutkulturen und eine Urinkultur angelegt sowie eine parasitologische Stuhluntersuchung veranlasst. Die Ergebnisse waren allesamt ohne Erregernachweis. Ebenso verblieben alle infektiologisch relevanten Serologien (HIV, Hepatitis B/C, Lues-Screening, Quantiferontest) negativ. Ein Urinsediment war ohne Erythrozyturie, zeigte jedoch eine prärenale Proteinurie mit Hinweisen einer tubulären Schädigung. Die Nierenfunktion lag mit einer eGFR (CKD-EPI2009) mit 85 ml/min im Normalbereich. Die extern veranlassten Hautbiopsien demonstrierten das Bild einer Kleingefäss-vaskulitis mit ausgeprägter Eosinophilie und fibrinoiden Gefässnekrosen. Eine zwischenzeitlich durchgeführte Biopsie aus der unteren Nasenmuschel zeigte keinen Hinweis für eine Vaskulitis, Granulome oder eosinophile Entzündung. Eine initial geplante diagnostische Bronchoskopie wurde bei fehlender therapeutischer Konsequenz abgesagt. In einer TTE/TEE-Untersuchung ergab sich kein Hinweis für eine kardiale Manifestation der vermuteten Grunderkrankung. In einer ergänzenden Abdomensonographie ergab sich kein Anhalt einer abdominalen Organmanifestation, Lymphadenopathie oder Hepatosplenomegalie.
Diagnose
In der Zusammenschau der Befunde und nach Ausschluss relevanter Differenzialdiagnosen wurde eine eosinophile Granulomatose mit Polyangiitis diagnostiziert.
Kommentar über Therapie und Verlauf
Aufgrund von persistierenden Fieberepisoden und progredienter Purpura wurde bei hochgradigem Verdacht einer EGPA bereits vor Erhalt der Biopsieergebnisse mit einer Hochdosis-Steroidtherapie (Methylprednison 500mg/24h intravenös über 3 Tage) unter der prophylaktischen Therapie mit einem Protonenpumpeninhibitor und der Gabe von Calcium/Vitamin D3 begonnen. Anschliessend wurde eine orale Steroidtherapie mit Prednison 1mg/kg Idealgewicht fortgeführt (4). Bei langfristiger Prednisontherapie mit einer Tagesdosis von über 20 mg (> 4 Wochen) initiierten wir zusätzlich eine Pneumocystis jirovecii (PJP)-Prophylaxe mit Sulfamethoxazol/Trimethoprim. Nach definitiver Diagnosestellung und bei Fehlen von schlechten Prognosefaktoren planten wir im weiteren Verlauf eine Antikörpertherapie mit Mepolizumab.
Unter der Steroidtherapie kam es bei kardialer Vorerkrankung zu einer rechtsführenden kardialen Dekompensation, sodass eine Negativbilanzierung mittels Schleifendiuretikum etabliert werden musste. Die bisherige Herzinsuffizienztherapie wurde um einen Angiotensin-II-Rezeptorantagonisten und einen SGLT2-Inhibitor ergänzt.
Unter der etablierten Therapie sahen wir eine rasche Normalisierung der Bluteosinophilie und der humoralen Entzündungsaktivität. Klinisch blasste die Purpura vollständig ab, weitere Fieberepisoden traten nicht auf. In der CT-graphischen Kontrolle ca. 2 Monate nach Therapieeinleitung waren die Groundglass-Opazitäten und bronchiolitischen Veränderungen vollständig regredient.
Wir beschreiben hier einen 61-jährigen Patienten mit langjährig bestehendem eosinophilen Asthma und einer Erstmanifestation einer eosinophilen Granulomatose mit Polyangiitis. Es handelt sich um eine perakute, potenziell lebensbedrohliche Erkrankung mit diversen möglichen Organmanifestationen. Die Diagnosestellung erfordert aufgrund verschiedener Differenzialdiagnosen aus dem eosinophilen und rheumatischen Formenkreis eine enge Zusammenarbeit mehrerer Fachdisziplinen und setzt sich aus Elementen der klinischen Untersuchung, Laboranalytik, Funktionsdiagnostik, Bildgebung und Histopathologie zusammen.
Das Therapieschema ist abhängig vom Schweregrad der Erkrankung. Dieser kann z.B. mittels Five-factor Score (Tab. 2) eingeschätzt werden (8). Die Induktionstherapie besteht aus Glucocorticoiden, welche bei schwerer Erkrankung mit Cyclophosphamid oder Rituximab kombiniert werden müssen. Mittels Glucocorticoid-Monotherapie konnte analog Ribi et. al (9) bei Fehlen von schlechten Prognosefaktoren in 93 % der Fälle eine Remission induziert werden, jedoch kam es hierbei zu einer Rezidivrate von 35 % im ersten Jahr. Durch eine Kombinationstherapie mit dem monoklonalen Interleukin-5-Antikörper Mepolizumab konnte eine signifikant längere Remissionsdauer mit konsekutiver Glucocorticoid-Einsparung erzielt werden (6). Alternativ ist eine Kombination von Glucocorticoiden und Azathioprin, Methotrexat oder Mycophenolat möglich. Die Behandlung mit Cyclophosphamid oder B-Zelldepletion ist schweren, organ- bzw. lebensgefährdenden Verläufen vorbehalten.

Abkürzungen
ABPA Allergische bronchopulmonale Aspergillose
ACR/EULAR American College of Rheumatology / European League Against Rheumatism
ANCA Antineutrophile-cytoplasmatische Antikörper
CT Computertomographie
CKD-EPI Chronic Kidney Disease Epidemiology Collaboration
eGFR Estimated Glomerular Filtration Rate
EGPA Eosinophile Granulomatose mit Polyangiitis
FEV1 Forced Exspiratory Volume in one Second
FISH Fluorescence in Situ Hybridization
FIP1L1-PDGFRA FIP1-like1-platelet-derived Growth Factor Receptor ∂
FVC Forced Vital Capacity
G/l Giga pro Liter
HES Hypereosinophiles Syndrom
HIV Human Immunodeficiency Virus
IgE Immunglobulin E
IgG Immunglobulin G
PCR Polymerase Chain Reaction
PJP Pneumocystis-jirovecii-Pneumonie
SGLT2 Sodium Glucose Transporter 2
ZNS Zentrales Nervensystem
Klinik für Pneumologie
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
silvia.ulrich@usz.ch
1. Salahuddin M et al. Pulmonary Eosinophilia. In: StatPearls (Internet). Treasure Island (FL): StatPearls Publishing; 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470600/
2. Rosenberg CE et al. Approach to Eosinophilia Presenting With Pulmonary Symptoms. Chest. 2021 doi: 10.1016/j.chest.2020.09.247.Cottin V. Eosinophilic Lung Diseases. Immunol Allergy Clin North Am. 2023. doi: 10.1016/j.iac.2023.01.002.
3. Chung SA et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Care Res (Hoboken). 2021;73(8):1088-1105. doi: 10.1002/acr.24634.
4. Holle JU et al. Eosinophilie: hypereosinophiles Syndrom versus eosinophile Granulomatose mit Polyangiitis. Z Rheumatol. 2023;82(4):307-320. German. doi: 10.1007/s00393-023-01345-2.
5. Wechsler ME et. Al. EGPA Mepolizumab Study Team. Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N Engl J Med. 2017. doi: 10.1056/NEJMoa1702079.
6. Emmi G et al. Evidence-Based Guideline for the diagnosis and management of eosinophilic granulomatosis with polyangiitis. Nat Rev Rheumatol. 2023;19(6):378-393. doi: 10.1038/s41584-023-00958-w.
7. Guillevin L et al. French Vasculitis Study Group (FVSG). The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore). 2011;90(1):19-27. doi: 10.1097/MD.0b013e318205a4c6.
8. Ribi C et. al. French Vasculitis Study Group. Treatment of Churg-Strauss syndrome without poor-prognosis factors: a multicenter, prospective, randomized, open-label study of seventy-two patients. Arthritis Rheum. 2008. doi: 10.1002/art.23198.

PRAXIS
- Vol. 114
- Ausgabe 1
- Januar 2025