Das kolorektale Karzinom (KRK) ist in der Schweiz hinsichtlich jährlicher Neuerkrankungen und Krebstodesfällen die dritthäufigste Karzinomart. Da die meisten Kantone ein organisiertes Vorsorgeprogramm durchführen, werden vermehrt Personen mit einer positiven Familienanamnese für das KRK erfasst. In der Mehrheit liegt die sog. familiäre Form des KRK vor, eine erbliche Form im engeren Sinne ist viel weniger häufig. Verwandte von Patienten mit einem KRK sind bezüglich Risiko, an einem KRK zu erkranken, eine heterogene Gruppe. Eine möglichst gute Einschätzung des Erkrankungsrisikos kann das Nutzen-Risiko-Verhältnis einer intensivierten Vorsorge optimieren. Diese Empfehlungen («Expert Opinion Statement») sollen im klinischen Alltag als Grundlage dienen für die Planung der Vorsorge, Überwachung und humangenetischen Beratung bei Vorliegen einer für das KRK positiven Familienanamnese.
Schlüsselwörter: familiäres Kolorektalkarzinom, erbliches Kolorektalkarzinom, Lynch-Syndrom, familiäre adenomatöse Polypose, MUTYH-assoziierte Polypose, Serratiertes Polypose-Syndrom
Einleitung
Das kolorektale Karzinom (KRK) ist in der Schweiz bei beiden Geschlechtern sowohl hinsichtlich jährlicher Neuerkrankungen wie Krebstodesfällen die dritthäufigste Karzinomart (1). Im Laufe des Lebens erkranken rund 3.7 % der Frauen und 5.2 % der Männer an einem KRK. Die Inzidenzraten blieben in der Schweiz in den letzten 30 Jahren weitgehend stabil, während die Mortalitätsraten rückläufig sind: aktuell liegt die 5-Jahres-Überlebensrate bei beiden Geschlechtern zwischen 65 und 70 %.
Ätiologisch kann zwischen dem sporadischen (sKRK), dem familiären (fKRK) und dem erblichen (eKRK) im engeren Sinne unterschieden werden mit je etwa 75 %, 20 % bzw. 5 % aller Neuerkrankungen (2). Der wichtigste Risikofaktor des sKRK ist das Alter, allerdings tritt diese Form in den letzten Dekaden immer häufiger schon vor dem 50. Lebensjahr auf, die Ursachen hierfür sind nur unvollständig verstanden (3). Zahlreiche Beobachtungsstudien dokumentieren ein erhöhtes Erkrankungsrisiko für das KRK bei einer positiven Familienanamnese (4, 5). In der Mehrheit dieser Fälle liegt die familiäre Form des KRK (fKRK) vor, bei der keine pathogene Keimbahnmutation in einem definierten Gen nachweisbar ist. Dem fKRK liegen vermutlich mono-, poly- sowie epigenetische Ursachen und Veränderungen im Mikrobiom zugrunde, deren Risiko, wie bei den anderen Formen, durch Umwelt- und Lebensstilfaktoren wie Ernährung, Rauchen, Alkoholkonsum, körperliche Aktivität und Gewicht etc. moduliert wird (6). Eine erbliche Form des KRK liegt bei einer positiven Familienanamnese nur selten vor, Angehörige betroffener Familien haben ein hohes Erkrankungsrisiko (2). Zu den erblichen (hereditären) Formen zählen insbesondere das eKRK ohne Polypose («nonpolyposis colorectal cancer», HNPCC, heute Lynch-Syndrom [LS] genannt) und verschiedene Polypose-Syndrome (siehe unten).
In der Schweiz ist die Darmkrebsvorsorge für Erwachsene zwischen dem 50. und 69. Lebensjahr mit normalem Erkrankungsrisiko mittels Koloskopie alle 10 Jahre oder durch quantitativen immunologischen Nachweis von okkultem Blut im Stuhl (FIT Test) alle zwei im Krankenversicherungsgesetz anerkannt. Die meisten Kantone führen ein organisiertes Screening-Programm durch. Dadurch werden vermehrt Personen erfasst und untersucht, welche über eine positive Familienanamnese für das KRK berichten. Diverse Fachgesellschaften empfehlen für Angehörige betroffener Familien eine intensivierte Vorsorge (7, 8, 9, 10). Für die Teilnahme an der Vorsorge müssen individuelle Vor- und Nachteile, aber auch gesellschaftliche Faktoren wie Kosten, limitierte personelle Ressourcen etc. berücksichtigt werden. Eine möglichst gute Einschätzung des Erkrankungsrisikos kann das Nutzen-Risiko-Verhältnis der ggf. intensivierten Vorsorge optimieren (11).
Es handelt sich hier um Empfehlungen im Sinne eines sog. Expert Opinion Statement. Diese ersten Schweizer Empfehlungen für das Vorgehen bei einer für das KRK positiven Familienanamnese sollen im klinischen Alltag als pragmatische Grundlage für die Planung der Vorsorge und Überwachung sowie der humangenetischen Beratung dienen. Im Rahmen eines mehrstufigen interdisziplinären Prozesses wurde dieses «Expert Opinion Statement» im Auftrag der Schweizerischen Gesellschaft für Gastroenterologie und Hepatologie durch die von den beteiligten Fachgesellschaften benannten Fachleute erarbeitet und repräsentiert eine schweizerische Perspektive. Deren Anwendbarkeit soll im Einzelfall geprüft und der individuellen Situation der Patientinnen und Patienten unter Berücksichtigung der gesamten klinischen Situation angepasst werden. Daten aus randomisierten kontrollierten Studien gibt es kaum, d.h., die verfügbare Evidenz für diese Empfehlungen ist von moderater, teilweise auch nur niedriger Evidenz.
Familiärer Darmkrebs
Allgemein
Rund 20–30 % aller Patienten mit einem KRK haben Verwandte mit dem gleichen Tumorleiden, wobei in rund der Hälfte ein Familienmitglied ersten Grades (Eltern, Geschwister, Kinder) betroffen ist (2). Beim fKRK finden sich oft mehrere, manchmal auch vor dem 50. Lebensjahr erkrankte Angehörige. Exom-Sequenzierungen bei mehr als 3000 Patienten mit einem fKRK konnten in ca. 16 % eine pathogene Keimbahnvariante in einem der bislang bekannten Prädispositionsgene identifizieren (12).
Das fKRK ist eine heterogene Gruppe, deren relative Risikoerhöhung durch den Verwandtschaftsgrad, die Anzahl betroffener Familienmitglieder und deren Erkrankungsalter beeinflusst wird (13, 14). Gemäss Beobachtungsstudien ist das Erkrankungsrisiko für Personen betroffener Familien etwa 2–6-fach erhöht im Vergleich zur Allgemeinbevölkerung (4). Beim Entscheid für das Screening soll auch das absolute Erkrankungsrisiko berücksichtigt werden (14, 15). Anhand der oben erwähnten Zahlen für die Schweiz ergibt sich ein Lebenszeitrisiko etwa zwischen 8 und 20 %. Am höchsten ist das Risiko, wenn Familienmitglieder mit Verwandtschaft im 1. Grad eine KRK-Diagnose hatten und die Erkrankung vor dem 50. Lebensjahr aufgetreten ist (13, 14).
Die möglichst gute Abschätzung der Risikoerhöhung durch eine detaillierte Erhebung der Familienanamnese ist demnach für die weitere Planung einer sinnvollen und ggf. intensivierten Vorsorgestrategie (welche Methode ab welchem Alter, Häufigkeit der Testung) der gesunden Familienmitglieder wichtig. Allerdings ist die Familienanamnese für das KRK und Polypen nur beschränkt zuverlässig, da diese den Angehörigen oft nicht oder nur unvollständig bekannt ist (16). Hinzu kommt, dass Familien heutzutage immer kleiner werden, was die Erkennung eines hereditären Syndroms erschweren kann. Die Familienanamnese verändert sich mit der Zeit, daher soll diese periodisch neu erhoben werden.
Vorsorge
Die bestehenden Richtlinien basieren auf der aktuell verfügbaren Literatur. Evidenz aus prospektiven randomisierten Studien gibt es nicht, die zugrunde liegenden Daten stammen aus Beobachtungsstudien, deren Qualität höchstens moderat ist und Spielraum für unterschiedliche Interpretationen zulässt. Grundlage für die Anwendung einer fKRK-Screening-Strategie ist, dass eine erbliche Form eines KRK weitgehend ausgeschlossen oder sehr unwahrscheinlich ist (siehe Kapitel: Erblicher Darmkrebs). Wie erwähnt, ist das Erkrankungsrisiko beim fKRK sehr variabel. Da Familienangehörige im Verwandtschaftsgrad 2 (Enkel/-in, Grosseltern, Onkel/Tante) kaum und weiter entfernte Verwandte kein erhöhtes Erkrankungsrisiko haben, empfehlen die meisten Fachgesellschaften bei dieser Konstellation keine intensivierte Vorsorge, sondern die Teilnahme an den lokalen Screening-Programmen für Personen mit Durchschnittsrisiko. Da hingegen Personen im Verwandtschaftsgrad 1 ein im Vergleich zur Allgemeinbevölkerung erhöhtes Darmkrebsrisiko haben, erscheint für diese Personen eine intensivierte Vorsorge gerechtfertigt, auch wenn der protektive Effekt der Screening-Koloskopie in dieser Gruppe nur durch wenige Beobachtungsstudien untermauert ist (17, 18).
Je jünger die betroffene Person bei der Darmkrebsdiagnose war, umso höher ist das Risiko für Angehörige im Verwandtschaftsgrad 1 (14). Die Kriterien für die Klassifikation eines gegenüber der Allgemeinbevölkerung erhöhten KRK-Risikos sind bei den verschiedenen Fachgesellschaften sehr unterschiedlich. Historisch von Bedeutung waren die Amsterdam-Kriterien I (1991) bzw. II (1999), die zur Identifikation der dem Lynch-Syndrom (LS) zugrunde liegenden Gene bzw. der Erfassung von LS-Patient/-innen entwickelt wurden, sowie die revidierten Bethesda-Guidelines (2004), die mittels Untersuchung der Mikrosatelliteninstabilität im Tumor die Sensitivität weiter erhöht haben (für eine Kriterienzusammenstellung s. a. Jasperson et al., 2010) (2). Die Guidelines der «U.S. Multi-Society Task Force on Colorectal Cancer» (USMSTF) empfehlen eine intensivierte Vorsorge generell, wenn eine Person mit Verwandtschaftsgrad 1 ein KRK hatte, d.h. unabhängig vom Erkrankungsalter (7). Die «European Society of Gastrointestinal Endoscopy» (ESGE) und die «British Society of Gastroenterology» (BSG) empfehlen eine intensivierte Vorsorge bei nur einer betroffenen Person im Verwandtschaftsgrad 1, wenn deren Erkrankung vor dem 50. Lebensjahr auftrat, das «Cancer Council Australia» (CCA) bei Manifestation des KRK vor dem 60. Lebensjahr (Tab. 1) (8, 9, 10). Sind hingegen zwei Personen im Verwandtschaftsgrad 1 an einem KRK erkrankt, dann schlagen die meisten Fachgesellschaften eine intensivierte Vorsorge unabhängig von deren Erkrankungsalter vor.
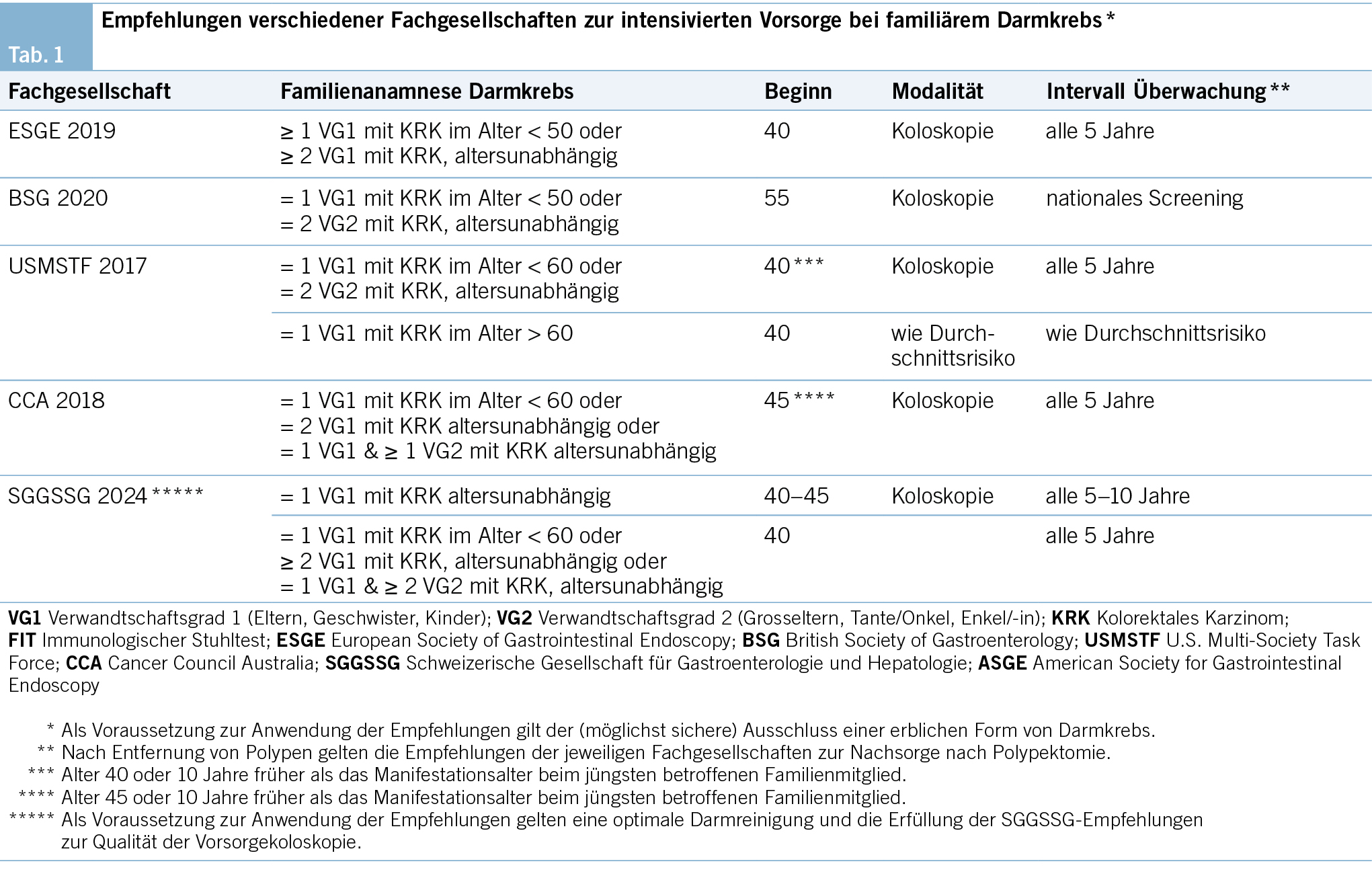
Es bestehen zwischen den verschiedenen Fachgesellschaften nicht nur verschiedene Definitionen zur Rechtfertigung der intensivierten Vorsorge, sondern auch unterschiedliche Vorgehensweisen, ab welchem Alter und mit welcher Häufigkeit die Vorsorge erfolgen soll (7, 8, 9, 10). Wie in Tab. 1 dargestellt, variiert der Beginn der Vorsorge bei fKRK je nach Fachgesellschaft zwischen 40 und 55 Jahren. Wenn eine intensivierte Vorsorge empfohlen ist, dann soll diese mittels Koloskopie erfolgen (10). Wenn die Koloskopie abgelehnt wird oder nicht durchführbar ist, dann kann alternativ die Vorsorge mit FIT Test erfolgen, vorzugsweise jährlich (19). Das weitere Vorgehen richtet sich nach der Konstellation der Familienanamnese (Tab. 1), dem Befund der Erstkoloskopie bzw. nach endoskopischer Entfernung von Polypen entsprechend den Richtlinien der verschiedenen Fachgesellschaften zur Nachsorge nach Polypektomie (20). Eine kürzlich publizierte Übersichtsarbeit fasst die Guidelines dieser und weiterer Fachgesellschaften zusammen und stellt die Divergenzen dar (21).
Nur wenige Studien haben untersucht, ob nach endoskopischer Polypektomie das Risiko für metachrone Polypen höher ist bei positiver KRK-Familienanamnese im Vergleich zu unauffälliger Familiengeschichte. Für die Entwicklung von adenomatösen Polypen (AP) konnte dies gezeigt werden, dabei war die Risikoerhöhung etwa gleich hoch für metachrone fortgeschrittene wie nicht fortgeschrittene AP. Die Risikoerhöhung war grösser für jüngere (< 50) im Vergleich mit älteren Personen und wenn mindestens zwei erstgradig Verwandte ein KRK hatten (22, 23). Die ESGE und BSG empfehlen dennoch die gleichen Überwachungsintervalle nach endoskopischer Polypenentfernung für Personen mit positiver KRK-Familienanamnese wie für Personen mit Durchschnittsrisiko.
Empfehlungen
Anhand der aktuellen Datenlage und der Guidelines anderer Fachgesellschaften empfehlen wir das intensivierte Screening mittels Koloskopie alle 5 Jahre ab Alter 40 für Personen, bei denen ein Familienangehöriger im Verwandtschaftsgrad 1 vor dem 60. Lebensjahr an einem KRK erkrankt ist, für Personen mit zwei oder mehr betroffenen Angehörigen derselben Familienseite im Verwandtschaftsgrad 1, unabhängig von deren Erkrankungsalter, sowie für Personen, bei denen ein Familienangehöriger im Verwandtschaftsgrad 1 und zusätzlich mindestens zwei Angehörige mit Verwandtschaftsgrad 2 betroffen sind. Die Nachsorge nach koloskopischer Polypektomie kann entsprechend den revidierten schweizerischen Konsensusempfehlungen erfolgen bzw. im Einzelfall, möglicherweise bei jüngeren Betroffenen, verkürzt werden (20).
Nebst dem möglichst sicheren Ausschluss einer erblichen Form eines KRK beim Indexpatienten ist die hohe Qualität der Erstkoloskopie Voraussetzung für die Anwendung dieser Empfehlungen. Die Koloskopie soll mit High-definition-Auflösung erfolgen, hingegen gibt es keine Evidenz für den Nutzen der generellen Anwendung der Chromoendoskopie (8). Letztlich sei erwähnt, dass Angehörige von Familien mit irgendeiner Form des familiären Auftretens eines KRK auf die Relevanz der Primärprävention hingewiesen werden sollen (normaler BMI, nicht rauchen, regelmässige körperliche Aktivität, moderater Konsum von rotem und prozessiertem Fleisch sowie Alkohol) (24).
Verwandte von Patienten mit kolorektalen Polypen
Nur wenige Fachgesellschaften haben Guidelines zur Gestaltung der Vorsorge bei Familienangehörigen von Personen mit gutartigen kolorektalen Polypen publiziert. Die verfügbare Datenlage hierzu ist kontrovers (25, 26). Die Risikoerhöhung für das Auftreten eines KRK scheint abhängig vom Alter des betroffenen Familienmitgliedes zu sein und ob bei diesem nicht fortgeschrittene oder fortgeschrittene Polypen vorgelegen haben. Eine intensivierte Vorsorge wird nur empfohlen, wenn bei einem Verwandten im Verwandtschaftsgrad 1 fortgeschrittene AP abgetragen wurden (fortgeschrittene AP: ≥ 10 mm oder Nachweis von hochgradiger Dysplasie oder ≥ 5 AP unabhängig von Grösse und Dysplasiegrad; fortgeschrittene serratierte Polypen (SP): ≥ 10 mm oder Nachweis irgendeiner Dysplasie oder ≥ 5 SP unabhängig von Grösse und Dysplasie, traditionell serratierte Adenome unabhängig von Grösse und Dysplasie). Ist die Histologie der entfernten Polypen nicht bekannt, dann wird von einem nicht fortgeschrittenen Polypen ausgegangen und keine intensivierte Vorsorge empfohlen. Weitgehend unklar ist die Datenlage nach Abtragung von fortgeschrittenen SP: hier empfiehlt die USMSTF das gleiche Vorgehen wie bei fortgeschrittenen AP, während mehrere andere Fachgesellschaften hierzu keine spezifischen Empfehlungen abgeben (7, 8, 9, 10). Demgegenüber ergab eine grosse schwedische Studie ein erhöhtes KRK-Risiko für erstgradige Verwandte von Familienmitgliedern mit kolorektalen Polypen, und zwar unabhängig von deren Histologie (27). Dabei zeigte sich vor allem ein zunehmendes Risiko für das Auftreten eines KRK vor dem 50. Lebensjahr, wenn mehrere Familienmitglieder Polypen hatten und je jünger deren Manifestationsalter war. Sollten sich diese Daten bestätigen, dann wird möglicherweise in Zukunft eine intensivierte Vorsorge bei einer positiven Familienanamnese für Polypen empfohlen, sofern mindestens zwei Familienangehörige im Verwandtschaftsgrad 1 Polypen vor dem 50. Lebensjahr hatten, unabhängig von deren Histologie, welche oft nicht verfügbar ist.
Empfehlungen
Anhand der derzeit verfügbaren Datenlage und den Guidelines anderer Fachgesellschaften empfehlen wir das Screening mittels Koloskopie alle 5–10 Jahre ab Alter 40 für Personen mit einem Familienangehörigen im Verwandtschaftsgrad 1, bei dem vor dem 50. Lebensjahr ein dokumentierter fortgeschrittener Polyp (AP oder SP) abgetragen wurde. Anhand der derzeitigen Datenlage können keine Empfehlungen gemacht werden bei einer anderweitig positiven Familienanamnese für kolorektale Polypen.
Serratiertes Polypose-Syndrom
Das serratierte Polypose-Syndrom (SPS) ist das häufigste kolorektale Polypose-Syndrom mit einer Prävalenz von etwa 1:240 (28). Eine 2022 publizierte Metaanalyse berichtete über ein KRK-Risiko für SPS-Patienten von 20 % (29). Die Mehrheit der Karzinome wurde zum Zeitpunkt der SPS-Diagnose gestellt, das Karzinomrisiko während der Überwachung lag bei knapp 3 %. Die dem SPS zugrunde liegenden molekularen Mechanismen sind weitgehend unbekannt, das SPS ist daher klinisch definiert: (i) ≥ 5 serratierte Polypen (SP) proximal des Rektums, alle ≥ 5 mm, zwei dieser Polypen müssen ≥ 10 mm sein; (ii) > 20 serratierte Polypen unabhängig von deren Grösse, ≥ 5 dieser Polypen müssen proximal des Rektums sein. Die Anzahl Polypen wird kumulativ über mehrere Koloskopien berechnet und umfasst alle SP-Subtypen (29). Auch wenn kein erbliches Syndrom im engeren Sinne vorliegt, haben erstgradige Verwandte von Patienten mit SPS ein etwa 5-fach erhöhtes KRK-Risiko (30).
Die Überwachung von SPS-Patienten mittels Koloskopie kann alle 1–2 Jahre erfolgen (9). Einige Fachgesellschaften empfehlen zunächst eine Clearing-Phase mit Entfernung aller Polypen (ausser HP < 5 mm), gefolgt von jährlicher Überwachung bei mindestens einem fortgeschrittenen Polypen (Adenom oder serratierter Polyp) oder ≥ 5 nicht fortgeschrittenen Polypen bzw. 2-jährlicher Überwachung ohne Nachweis einer solchen Konstellation (31, 32). Für Verwandte im ersten Verwandtschaftsgrad von SPS-Patienten kann eine koloskopische Überwachung alle 5 Jahre ab Alter 40–45 (oder SPS-Manifestationsalter des erstgradig Verwandten oder 10 Jahre früher als dessen Erkrankungsalter) erfolgen (9, 33).
Empfehlung
Anhand der derzeit verfügbaren Datenlage schlagen wir das genannte Phänotyp-abhängige Vorgehen mit einer Clearing-Phase gefolgt von 2-jährlichen Koloskopien vor, sofern keine fortgeschrittenen und weniger als 5 Polypen vorliegen (grössenunabhängig, alle Subtypen). Falls dies jedoch der Fall ist, dann soll die koloskopische Kontrolle jährlich erfolgen.
Familiäres Kolorektales Karzinom Typ X
Diese Entität umfasst die rund 40 % Patienten mit klinischem Verdacht auf das Vorliegen eines LS (3 Verwandte im Verwandtschaftsgrad 1 mit KRK, 2 Generationen betroffen), ohne dass eine MMR-Defizienz oder Mutation in einem der MMR-Gene nachweisbar ist. Das Karzinomrisiko in dieser Gruppe beschränkt sich auf das Kolorektum und ist kleiner als beim LS und Lynch-like Syndrom (LLS) (34), sodass meist eine Koloskopie alle 3–5 Jahre ab Alter 40 (oder 10 Jahre früher als das jüngste Manifestationsalter innerhalb der Familie) vorgeschlagen wird (9, 35).
Empfehlung
Anhand der aktuellen Datenlage und der Guidelines anderer Fachgesellschaften schlagen wir folgendes Screening vor: Koloskopie alle 3–5 Jahre ab Alter 40 oder 10 Jahre früher als das Erkrankungsalter des jüngsten betroffenen Familienmitgliedes.
Erblicher Darmkrebs
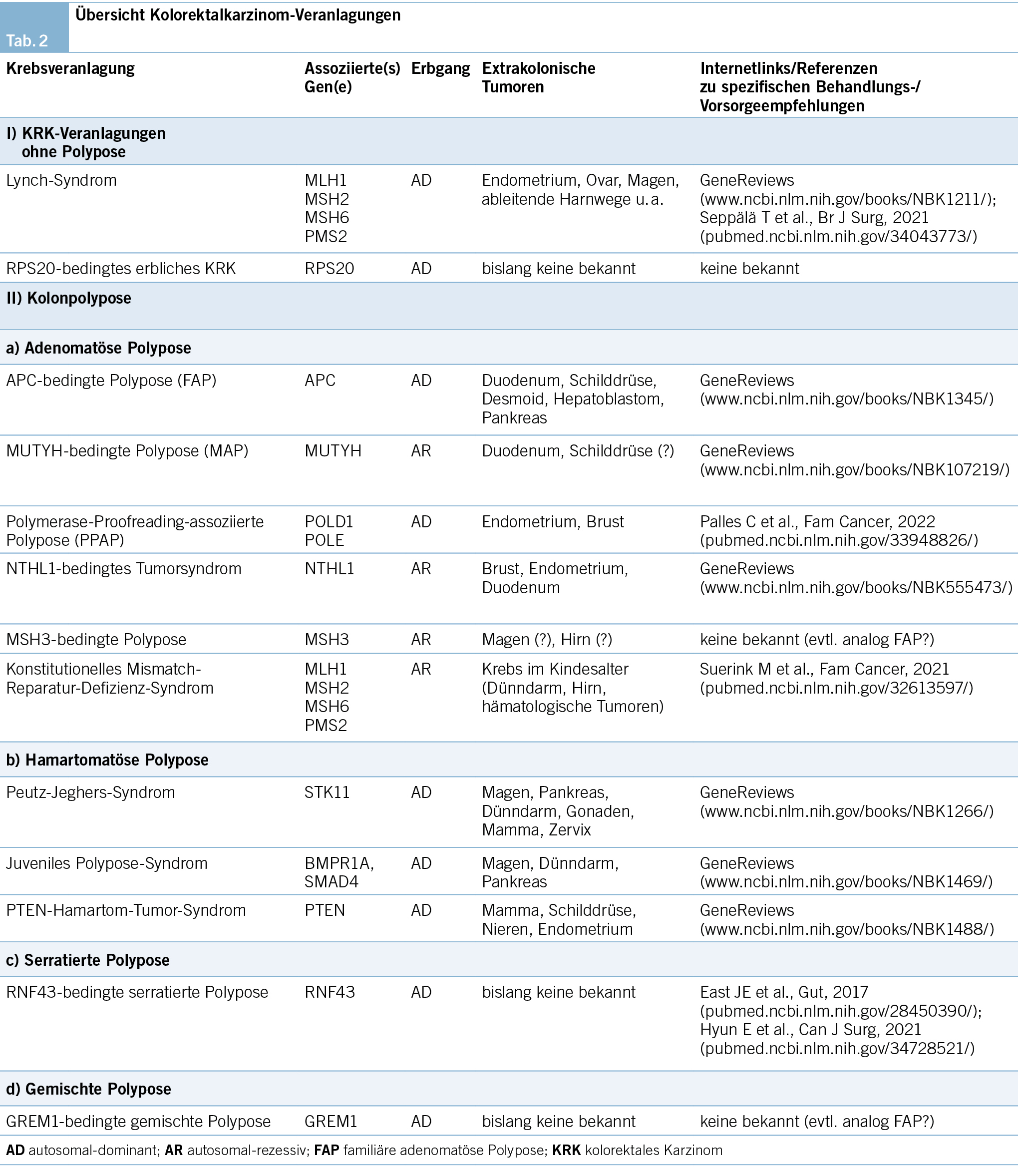
Beim erblichen KRK kann zwischen Darmkrebs ohne vorbestehende Kolon-Polypose («nonpolyposis colorectal cancer») und den Polypose-Erkrankungen unterschieden werden (Tab. 2), bei letzteren zudem nach der vorherrschenden Polypenart. Wie aus Tab. 2 ersichtlich, beschränkt sich die erhöhte Tumoranfälligkeit meist nicht nur auf ein einzelnes Organsystem wie das Kolorektum. Oft zeigen sich sowohl inter- wie intrafamiliär phänotypische Unterschiede, denen wohl komplexe Interaktionen zwischen genspezifischen Unterschieden sowie Umwelt- und Lifestyle-Faktoren zugrunde liegen (36).
Lynch-Syndrom
Mit einer Prävalenz von ca. 1 auf 279 Personen stellt das autosomal-dominant erbliche LS die weltweit häufigste Tumorveranlagung dar (37). Rund 3 % aller KRK entstehen auf dem Boden einer LS-Veranlagung, bei der pathogene Keimbahnvarianten in den MMR-Genen MLH1, MSH2/EPCAM, MSH6 und PMS2 zugrunde liegen. Die daraus resultierende MMR-Defizienz lässt sich im Tumorgewebe indirekt in Form einer Mikrosatelliteninstabilität (MSI) bzw. dem Verlust der MMR-Proteinexpression nachweisen.
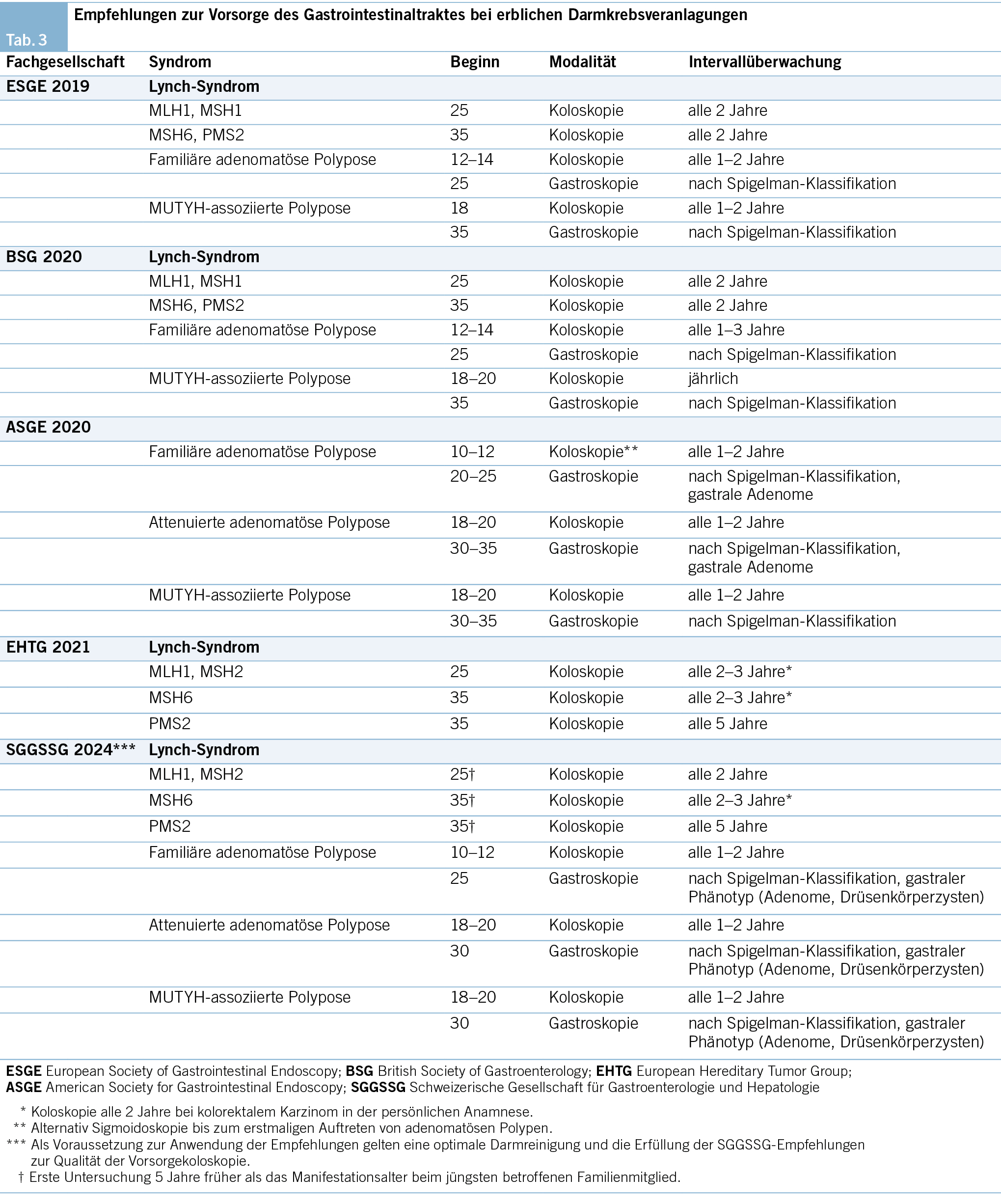
Prospektive Studien konnten ausgeprägte genspezifische Unterschiede für Tumorerkrankungen bei Personen mit nachgewiesener LS-Veranlagung aufzeigen: So beträgt das Lebenszeitrisiko für das KRK bei MLH1- und MSH2-Träger/-innen ca. 42–53 % und wird im Mittel um das 44. Lebensjahr diagnostiziert; MSH6- und PMS2-Träger/-innen entwickeln «lediglich» in ca. 3–20 % ein KRK erst mit 42–69 (MSH6) bzw. 61–66 Jahren (PMS2) (38, 39). Ähnlich verhält es sich mit anderen LS-assoziierten, extrakolonischen Tumorerkrankungen wie Endometrium- (MSH2, MSH6, MLH1: ca. 35–46 %; PMS2: ca. 13 %), Ovarial- (MSH2, MLH1, MSH6: ca. 11–17 %; PMS2: ca. 3 %) und Magen-/Dünndarmkarzinom (MSH2, MLH1: ca. 8–16 %; MSH6, PMS2: ca. 2–4 %) (40). Aufgrund dieser Unterschiede haben mehrere Fachgesellschaften ihre Empfehlungen für gynäkologische und gastroenterologische Vorsorgeuntersuchungen genspezifisch angepasst (Tab. 3) (41).
Bei Darmkrebspatienten und zur Vorsorgekoloskopie erscheinenden Personen mit positiver Familienanamnese für das KRK soll bei folgenden klinischen Konstellationen an ein LS gedacht werden: Darmkrebsdiagnose vor dem 50. Lebensjahr, syn- oder metachrone LS-assoziierte Tumoren unabhängig vom Erkrankungsalter, ein Angehöriger 1. oder 2. Grades mit vor dem 50. Lebensjahr diagnostiziertem, LS-assoziiertem Tumor bzw. 2 oder mehr Angehörige mit LS-assoziierten Tumoren unabhängig vom Erkrankungsalter. Weiter sollte eine genetische Abklärung bei Vorliegen einer auffälligen Familienanamnese, wie oben erwähnt, oder am Tumorgewebe nachgewiesener MMR-Defizienz oder somatischen MMR-Genalterationen in Betracht gezogen werden (37). Auch bei Vorliegen einer Lynch-Syndrom verdächtigen Klinik und im Tumor erhaltener MMR-Funktion sollte eine Abklärung der MMR-Gene in der Keimbahn diskutiert werden, da die diagnostische Sensitivität der erwähnten indirekten Methoden nur bei ca. 90 % liegt (42).
Immuncheckpoint-Inhibitoren werden heutzutage oft in der Behandlung von MMR-defizienten soliden Tumoren eingesetzt und daher alle neu diagnostizierten KRK (und zunehmend auch weitere Krebsarten) auf ihre MMR-Funktion überprüft. Dies ist nicht nur von grosser Bedeutung für die bessere Erfassung von LS-Patient/-innen, sondern eröffnet zudem neue Therapieoptionen bei ca. 10–15 % der Patient/-innen mit sporadischem MMR-defizientem KRK (43).
Die KRK-Vorsorge wird von verschiedenen Fachgesellschaften für Träger/-innen einer MLH1- oder MSH2-Mutation ab Alter 25 und bei MSH6- oder PMS2-Mutation ab Alter 35 empfohlen (Tab. 3). Die Vorsorge wird generell mittels Koloskopie und bei MLH1- oder MSH2-Trägerschaft alle 1–3 Jahre empfohlen, bei Mutation im MSH6-Gen alle 2–3 Jahre und bei einer Mutation im PMS2-Gen alle 5 Jahre. Prospektive erhobene Daten einer europäischen Studie zeigten hinsichtlich Inzidenz und Tumorstadium keinen Benefit bei jährlicher Koloskopie gegenüber einem weniger strikten Überwachungsprotokoll (44). Trotz Überwachung erreichte in dieser Studie die kumulative KRK-10-Jahresinzidenz bis zu 18 %. Dies bedeutet, dass durch die koloskopische Surveillance mit Polypektomie das KRK nicht immer verhindert, aber oft früher erkannt werden kann, mit entsprechend besserer Prognose. Die Gründe hierfür sind noch nicht vollständig geklärt (45). Gute Evidenz für den Nutzen einer regelmässigen Überwachung zur Senkung der Inzidenz und Mortalität von Karzinomen des Magens, Dünndarmes und Pankreas gibt es nicht.
Dementsprechend wird die Überwachung des oberen Gastrointestinaltraktes von den meisten Fachgesellschaften nicht routinemässig, sondern nur bei Vorliegen weiterer Risikofaktoren (familiäres Auftreten von Magenkarzinomen, Regionen mit hoher Inzidenz für Magenkrebs) empfohlen, meist ab Alter 30. Vorsorgeuntersuchungen für den Dünndarm und das Pankreas werden ebenso nicht generell, sondern nur auf individueller Basis empfohlen, bspw. bei (mehrfachem) familiärem Auftreten von Karzinomen in den entsprechenden Organen.
Empfehlungen
Anhand der aktuellen Datenlage und der Guidelines anderer Fachgesellschaften schlagen wir folgendes Screening vor (Tab. 3): Koloskopische Überwachung geschlechtsunabhängig alle 2 Jahre bei MLH1- oder MSH2-Mutation ab Alter 25, bei MSH6 ab Alter 35 alle 2–3 Jahre und bei PMS2-Mutation ab Alter 35 alle 5 Jahre. Beginn der Überwachung in allen Fällen wenigstens 5 Jahre früher als das Erkrankungsalter des jüngsten betroffenen Familienmitgliedes. Anpassung der Überwachung an den kolorektalen Phänotyp (Nachweis fortgeschrittener Polypen), zudem vorzeitige Untersuchung bei Auftreten von Symptomen. Obere Panendoskopie in Abhängigkeit des familiären Phänotyps (positive Familienanamnese für das Magenkarzinom). Wir empfehlen eine Helicobacter-Pylori-Diagnostik mit ggf. Eradikation. Den Patient/-innen soll ein gesunder Lebensstil empfohlen werden (wie die Vermeidung von Übergewicht, regelmässige Bewegung und eine ausgewogene Ernährung) (46).
Fortschritte in der Labordiagnostik und die Fortsetzung der prospektiven Datenerhebung in internationalen Konsortien wie der Prospective Lynch Syndrome Database (PLSD) (47) werden zu weiteren, klinisch relevanten Genotyp-Phänotyp-Korrelationen führen, welche Anpassungen des Screenings gastrointestinaler und anderer Organe erfordern. Daher sind in Tab. 2 diverse Webadressen aufgelistet, um sich nach den aktuellsten Vorsorgeuntersuchungen zu informieren. Der Vollständigkeit halber sei noch das autosomal-rezessiv erbliche, konstitutionelle Mismatch-Reparatur-Defizienz-Syndrom (CMMRD) erwähnt, eine seltene Krebsveranlagung des Kindesalters, die v. a. mit malignen hämatologischen, gastrointestinalen und ZNS-Tumoren einhergeht (40).
Lynch-like Syndrom
Das LLS beschreibt Personen mit MMR-defizientem KRK oder anderen LS-assoziierten Tumoren, ohne dass eine Keimbahnmutation in einem MMR-Gen nachweisbar ist (48). Da die Mehrheit aller KRK mit MSI nicht im Rahmen eines LS, sondern sporadisch als Folge einer Hypermethylierung im MLH1-Promoter auftreten, soll diese molekulare Aberration ausgeschlossen werden. Ist dies der Fall, dann ist in 50–70 % der Fälle die MMR-Defizienz durch (sporadisch aufgetretene) biallelische somatische MMR-Genmutationen erklärt. Um Betroffene und deren Verwandte nicht durch unnötige Überwachungen zu belasten, soll daher auch diese Konstellation im Labor ausgeschlossen werden (49, 50). Das KRK-Risiko in betroffenen Familien scheint kleiner als bei nachgewiesenem LS, aber höher als beim fKRK zu sein (34, 51). Die Datenlage zum wenig verstandenen LLS ist dürftig, vorgeschlagen wird beispielsweise die Überwachung mittels Koloskopie alle 2–3 Jahre für Betroffene und deren Verwandte im Verwandtschaftsgrad 1 ab Alter 25 bzw. in Abhängigkeit von der Familienanamnese (9, 52).
Empfehlung
Anhand der limitierten Evidenz können keine Empfehlungen gegeben werden. Die Überwachung soll individuell unter Berücksichtigung der Familienanamnese und des Phänotyps des/der betroffenen Patient/-in festgelegt werden.
Adenomatöse Polyposen
Die im Vergleich zum Lynch-Syndrom deutlich selteneren adenomatösen Polyposen-Syndrome sind für ca. 1 % der KRK insgesamt bzw. ca. 2 % aller vor dem 50. Lebensjahr diagnostizierten KRK verantwortlich. Diagnostisch unproblematisch ist die «klassische» Ausprägung mit Hunderten bis hin zu Tausenden von gastrointestinalen Adenomen (53). In bis zu einem Drittel liegt beim klassischen Phänotyp keine positive Familienanamnese vor, sodass von einer Neumutation ausgegangen wird. Liegen weniger als 100 Adenome vor, handelt es sich um eine attenuierte Polypose-Form, die genetisch heterogen ist und sowohl dem autosomal-dominanten (APC, POLD1, POLE) als auch dem autosomal-rezessiven Erbgang (MUTYH, NTHL1, MSH3, MBD4 u. a.) folgen kann.
Eine genetische Abklärung ist zu diskutieren, wenn bei einer Person kumulativ mindestens 10–20 Adenome nachgewiesen wurden. Wie Terlouw et al. gezeigt haben, liegt die Detektionswahrscheinlichkeit für pathogene APC- oder MUTYH-Varianten über 10 %, wenn bei einer Person vor dem 60. Lebensjahr kumulativ mehr als 10 bzw. vor dem 70. Lebensjahr mehr als 20 adenomatöse Polypen gefunden wurden (54). Weiter können das Vorliegen extrakolonischer Tumormanifestationen, wie z.B. Desmoid-Tumoren, multiple Osteome u. a., und eine auffällige Familiengeschichte (Angehöriger 1. Grades mit > 10 Adenomen) zusätzliche Hinweise auf eine hereditäre Polypose liefern. Das Risiko für die Entwicklung eines KRK nimmt bei der klassischen APC-bedingten familiären adenomatösen Polypose (FAP) bereits anfangs der zweiten Lebensdekade zu und erreicht ein Lebenszeitrisiko von beinahe 100 %. Bei Vorliegen der attenuierten Form (AFAP) treten Neoplasien etwas später und vor allem im rechten Hemikolon auf, das KRK-Risiko beträgt ca. 70 % (55, 56). Bei beiden Formen treten auch gehäuft Karzinome im oberen Gastrointestinaltrakt auf, im Bereich von Duodenum/Papilla Vateri in ca. 4–12 % und im Magen in ca. 1 %.
Die meisten Fachgesellschaften empfehlen bei nachgewiesener Trägerschaft einer autosomal-dominant erblichen APC-bedingten Kolonpolypose die 1–2-jährliche koloskopische Überwachung bei der klassischen Form ab dem Beginn und bei der attenuierten FAP (AFAP) ab dem Ende der zweiten Lebensdekade.
Bei den meisten Patienten mit APC-bedingter Polypose, besonders der klassischen Form (bis 90 %), finden sich im Magen zahlreiche Drüsenkörperzysten, Adenome sind weniger häufig, scheinen aber klinisch relevanter zu sein wegen des Risikos einer malignen Transformation. Die Strategien zur Überwachung im oberen Gastrointestinaltrakt bei FAP bzw. AFAP sind heterogen. Bei der klassischen Form wird eine obere Endoskopie mit Darstellung der Papilla Vateri ab dem 20.–25. Lebensjahr vorgeschlagen mit befundabhängiger (Spigelman-Klassifikation, Phänotyp im Magen) Wiederholung alle 6 Monate bis 5 Jahre. Da Träger/-innen einer pathogenen APC-Variante, die hinter APC-Kodon 1395 liegt, ein erhöhtes Lebenszeitrisiko (ca. 10–24 %) für Desmoid-Tumoren aufweisen, sollten bei diesen Personen mehrstufige Operationen vermieden und klinisch ein besonderes Augenmerk auf entsprechende abdominale Symptome gelegt werden, mit ggf. bildgebender Abklärung mittels MRI oder CT.
Bei der autosomal-rezessiven MUTYH-bedingten attenuierten Form der adenomatösen Polypose (MAP) liegt das Lebenszeitrisiko für ein KRK bei ca. 80 %, für ein Karzinom im Duodenum bei ca. 4 % und im Magen bei ca. 1 %. Wie bei der APC-assoziierten Form der attenuierten Polypose steigt das KRK-Risiko erst gegen Ende der zweiten Lebensdekade an, und auch die MAP manifestiert sich vor allem im rechten Hemikolon, dabei können nebst AP auch SP vorliegen. Dementsprechend empfehlen die meisten internationalen Fachgesellschaften für die Überwachung des Kolorektums bei MAP (nachgewiesene biallelische MUTYH-Träger/-innen), sofern die persönliche bzw. die Familienanamnese nicht auf einen besonderen Phänotyp hinweist, 1–2-jährliche Koloskopien ab dem 18.–20. Lebensjahr. Eine obere Endoskopie mit Darstellung der Papilla Vateri wird ab der 3. Dekade vorgeschlagen, wiederum befundabhängig alle 6 Monate bis 5 Jahre. Kontrovers wird derzeit diskutiert, ob monoallelische (heterozygote) MUTYH-Träger/-innen ein (etwa 2-fach) erhöhtes KRK-Risiko tragen. Teilweise wird empfohlen, ab dem 40. Lebensjahr etwa alle 5 Jahre eine Koloskopie durchzuführen, insbesondere wenn in der Familie ein erstgradig Verwandter an einem KRK erkrankte.
Empfehlungen
Anhand der aktuellen Datenlage und der Guidelines anderer Fachgesellschaften schlagen wir folgendes Screening vor (Tab. 3): Koloskopie alle 1–2 Jahre ab Alter 12–14 bei klassischer FAP, bei AFAP und MAP ab Alter 18–20. Gastroskopie mit Darstellung der Papilla Vateri alle 1–5 Jahre ab Alter 25. Anpassung der endoskopischen Überwachung des oberen und unteren Gastrointestinaltraktes an den Phänotyp und bei Auftreten von Symptomen. Bei Status nach Operation soll das Restrektum oder der Pouch ca. alle 6–12 Monate endoskopisch kontrolliert werden. In den letzten 20 Jahren wurden weitere, sehr seltene Non- bzw. Polypose-Formen entdeckt, die mit einem erhöhtem Risiko für das KRK und oft auch für extrakolonische Tumore einhergehen (Tab. 2). Aufgrund der Seltenheit wird in dieser Arbeit nicht näher auf diese eingegangen.
Genetische Beratung und Abklärung
Bei ca. 13 % (9 %–26 %) der KRK-Patient/-innen, deren Erkrankung vor dem 50. Lebensjahr diagnostiziert wurde, lässt sich eine pathogene Keimbahnvariante in einem der 17 bislang bekannten Gene identifizieren (57), was von klinischer Relevanz sein kann. Das frühzeitige Erkennen einer erblichen Darmkrebserkrankung hat dabei nicht nur Konsequenzen für das chirurgisch-onkologische Vorgehen und die Gestaltung der Nach- bzw. Vorsorge beim Betroffenen, sondern ist auch von essenzieller Bedeutung für dessen gesunde Familienangehörige. So ermöglicht dies in der Folge auch den Angehörigen (u. a. Eltern, Geschwister, Kinder meist erst ab 18. Lebensjahr), sich nach entsprechender genetischer Beratung und angemessener Bedenkzeit prädiktiv auf Trägerschaft testen zu lassen (Trägerwahrscheinlichkeit bei Verwandten 1. Grades: 25 % bzw. 50 %) und so für sich die Notwendigkeit regelmässiger Krebsvorsorgeuntersuchungen zu klären.
Die Kosten einer molekulargenetischen Abklärung von 1–10 Genen belaufen sich auf ca. CHF 3 000 – 4 000.– und sind als Pflichtleistungen in der Analysenliste (Anhang 3 der Krankenpflege-Leistungsverordnung) entweder spezifisch (Lynch-Syndrom, APC-bedingte Polypose) oder in genereller Form als «Seltene erbliche Tumorkrankheiten» aufgeführt. Für Letztere sollte zur Sicherung der Kostenbeteiligung vorgängig ein sog. Orphan Disease-Antrag beim vertrauensärztlichen Dienst des Krankenversicherers eingereicht werden (Antragsformular: https://sgmg.ch/de/fachthemen#fachthemen-dokumente).
Vor Veranlassung einer diagnostischen genetischen Abklärung bedarf es, wie im Bundesgesetz über genetische Untersuchungen beim Menschen (GUMG) festgehalten, einer hinreichenden Aufklärung, die der/die auftraggebende Arzt/Ärztin entweder selbst durchführt oder eine genetische Beratung veranlasst, sowie der Zustimmung des Patienten bzw. der Patientin.
Vor und nach einer präsymptomatischen (prädiktiven) genetischen Testung ist eine fachkundige genetische Beratung gesetzlich vorgeschrieben, in der nicht nur auf Aussagekraft, Grenzen und medizinische Konsequenzen einer Tragertestung bzw. dem Verzicht darauf eingegangen wird, sondern auch psychosoziale und (versicherungs)rechtliche Aspekte diskutiert werden. Die Trägertestung beläuft sich auf ca. CHF 400.– und stellt, mit Ausnahme von Lynch-Syndrom und APC-bedingter Polypose, keine Pflichtleistung der Krankenversicherer dar.
Chemoprävention
In diversen Labor- und klinischen Studien konnte ein protektiver Effekt diverser Substanzen (NSAR, Statine, Vitamine etc.) auf die Entstehung und Progression kolorektaler Neoplasien nachgewiesen werden (58). So zeigte sich für die am besten untersuchte Medikamentengruppe, NSAR inkl. Aspirin, dass bei FAP-Patienten die Anzahl und Grösse von Polypen und bei Patienten mit Lynch-Syndrom das KRK-Risiko gesenkt werden kann. In der CAPP-2-Studie wurde das KRK-Risiko nach rund 10 Jahren durch die Einnahme von 600 mg Aspirin täglich im Vergleich zu Placebo um rund 35 % gesenkt (59). Verschiedene Aspekte wie die optimale Dosierung und Dauer der Chemoprävention mit Aspirin sind aber noch ungenügend geklärt. Dementsprechend unterstützen einige Fachgesellschaften die prophylaktische Aspirin-Einnahme bei LS, während andere keine Stellungnahme abgeben. Risiko und Benefit müssen daher individuell abgeschätzt und auch im Rahmen allfälliger Komorbiditäten und des Alters beurteilt werden.

Abkürzungen
AFAP Attenuierte familiäre adenomatöse Polypose
AP Adenomatöser Polyp
BSG British Society of Gastroenterology
CCA Cancer Council Australia
eKRK Erbliches kolorektales Karzinom
FAP Familiäre adenomatöse Polypose
FDR First-degree Relatives
FIT Test Immunologischer Test auf okkultes Blut im Stuhl
fKRK Familiäres kolorektales Karzinom
ESGE European Society of Gastrointestinal Endoscopy
HNPCC Hereditary Nonpolyposis Colorectal Cancer
HP Hyperplastischer Polyp
KRK Kolorektales Karzinom
LLS Lynch-like Syndrom
LS Lynch-Syndrom
MAP MUTYH-assoziierte Polypose
MMR Mismatch Repair
SP Serratierter Polyp
SPS Serratiertes Polypose-Syndrom
USMSTF U. S. Multi-Society Task Force
Disclaimer
Diese Empfehlungen müssen in der Zukunft überarbeitet und angepasst werden, abhängig von neuen Studiendaten und technologischen Möglichkeiten sowie basierend auf Erfahrungen im klinischen Alltag. Diese Empfehlungen sollen als Orientierung in der klinischen Praxis dienen und nicht als universell gültige Regeln angewendet werden. Die klinische Situation kann eine Abweichung von den vorgeschlagenen Empfehlungen erfordern.
Klinik für Gastroenterologie und Hepatologie Universitätsspital Zürich
Rämistrasse 100, 8091 Zürich
k.truninger@hin.ch
Institut für Medizinische Genetik und Pathologie Universitätsspital Basel
Schönbeinstrasse 40
4031 Basel
karl.heinimann@usb.ch
Die Autorschaft keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Bundesamt für Statistik. Krebs in der Schweiz 2015-2019. Bern; 2022 Dec.
2. Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010 Jun;138(6):2044–58.
3. Sinicrope FA. Increasing Incidence of Early-Onset Colorectal Cancer. N Engl J Med. 2022 Apr;386(16):1547–58.
4. Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001 Oct;96(10):2992–3003.
5. Patel SG, Karlitz JJ, Yen T, Lieu CH, Boland CR. The rising tide of early-onset colorectal cancer: a comprehensive review of epidemiology, clinical features, biology, risk factors, prevention, and early detection. Lancet Gastroenterol Hepatol. 2022 Mar;7(3):262–74.
6. Lichtenstein P, Holm N V, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000 Jul;343(2):78–85.
7. Rex DK, Boland CR, Dominitz JA, Giardiello FM, Johnson DA, Kaltenbach T, et al. Colorectal Cancer Screening: Recommendations for Physicians and Patients From the U.S. Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2017 Jul;153(1):307–23.
8. van Leerdam ME, Roos VH, van Hooft JE, Balaguer F, Dekker E, Kaminski MF, et al. Endoscopic management of Lynch syndrome and of familial risk of colorectal cancer: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy. 2019 Nov;51(11):1082–93.
9. Monahan KJ, Bradshaw N, Dolwani S, Desouza B, Dunlop MG, East JE, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020 Mar;69(3):411–44.
10. Jenkins MA, Ait Ouakrim D, Boussioutas A, Hopper JL, Ee HC, Emery JD, et al. Revised Australian national guidelines for colorectal cancer screening: family history. Med J Aust. 2018 Nov;209(10):455–60.
11. Kastrinos F, Kupfer SS, Gupta S. Colorectal Cancer Risk Assessment and Precision Approaches to Screening: Brave New World or Worlds Apart? Gastroenterology. 2023 Apr;164(5):812–27.
12. Chubb D, Broderick P, Dobbins SE, Frampton M, Kinnersley B, Penegar S, et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat Commun. 2016 Jun;7:11883.
13. Fuchs CS, Giovannucci EL, Colditz GA, Hunter DJ, Speizer FE, Willett WC. A prospective study of family history and the risk of colorectal cancer. N Engl J Med. 1994 Dec;331(25):1669–74.
14. Roos VH, Mangas-Sanjuan C, Rodriguez-Girondo M, Medina-Prado L, Steyerberg EW, Bossuyt PMM, et al. Effects of Family History on Relative and Absolute Risks for Colorectal Cancer: A Systematic Review and Meta-Analysis. Clin Gastroenterol Hepatol. 2019 Dec;17(13):2657-2667.e9.
15. Butterworth AS, Higgins JPT, Pharoah P. Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer. 2006 Jan;42(2):216–27.
16. Alvarez MD, Quintana I, Terradas M, Mur P, Balaguer F, Valle L. The inherited and familial component of early-onset colorectal cancer [Internet]. Vol. 10, Cells. 2021. p. 710. Available from: https://www.mdpi.com/2073-4409/10/3/710
17. Brenner H, Chang-Claude J, Seiler CM, Hoffmeister M. Long-term risk of colorectal cancer after negative colonoscopy. J Clin Oncol. 2011 Oct;29(28):3761–7.
18. Samadder NJ, Pappas L, Boucherr KM, Smith KR, Hanson H, Fraser A, et al. Long-Term Colorectal Cancer Incidence After Negative Colonoscopy in the State of Utah: The Effect of Family History. Am J Gastroenterol. 2017 Sep;112(9):1439–47.
19. Davidson KW, Barry MJ, Mangione CM, Cabana M, Caughey AB, Davis EM, et al. Screening for Colorectal Cancer: US Preventive Services Task Force Recommendation Statement. JAMA. 2021 May;325(19):1965–77.
20. Truninger K, Lugli A, Koeberle D. Nachsorge nach koloskopischer Polypektomie und Therapie des kolorektalen Karzinoms. Swiss Medical Forum. 2022;22:349–55.
21. Mangas-Sanjuan C, Jover R. Familial colorectal cancer. Best Pract Res Clin Gastroenterol. 2022;58–59:101798.
22. Jacobs ET, Gupta S, Baron JA, Cross AJ, Lieberman DA, Murphy G, et al. Family history of colorectal cancer in first-degree relatives and metachronous colorectal adenoma. Am J Gastroenterol. 2018 Jun;113(6):899–905.
23. Park CH, Kim NH, Park JH, Park D Il, Sohn C Il, Jung YS. Impact of family history of colorectal cancer on age-specific prevalence of colorectal neoplasia. J Gastroenterol Hepatol. 2019 Mar;34(3):537–43.
24. Wang S, Yuan Z, Wang Y, Zhao X, Gao W, Li H, et al. Modifiable lifestyle factors have a larger contribution to colorectal neoplasms than family history. BMC Cancer. 2022 Oct;22(1):1051.
25. Cottet V, Pariente A, Nalet B, Lafon J, Milan C, Olschwang S, et al. Colonoscopic screening of first-degree relatives of patients with large adenomas: increased risk of colorectal tumors. Gastroenterology. 2007 Oct;133(4):1086–92.
26. Tuohy TMF, Rowe KG, Mineau GP, Pimentel R, Burt RW, Samadder NJ. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer. 2014 Jan;120(1):35–42.
27. Song M, Emilsson L, Roelstraete B, Ludvigsson J. Risk of colorectal cancer in first degree relatives in patients with colorectal polyps: nationwide case-control study in Sweden. BMJ 2021; 373: n877.
28. Carballal S, Balaguer F, IJspeert JEG. Serrated polyposis syndrome; epidemiology and management. Best Practice and Research Clinical Gastroenterology 2022; 58-59: 101791.
29. Muller C, Yamada A, Ikegami S, Haider H, Komaki Y, Komaki F, et al. Risk of colorectal cancer in serrated polyposis syndrome: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2022; 20: 622-30.
30. Kanth P, Yu Z, Keener MB, Koptiuch C, Kohlmann WK, Neklason DW, et al. Cancer Risk in Patients With and Relatives of Serrated Polyposis Syndrome and Sporadic Sessile Serrated Lesions. Am J Gastroenterol. 2022 Feb;117(2):336–42.
31. Dekker E, Bleijenberg A, Balaguer F. Update on the World Health Organization Criteria for Diagnosis of Serrated Polyposis Syndrome. Vol. 158, Gastroenterology. United States; 2020. p. 1520–3.
32. Mankaney G, Rouphael C, Burke CA. Serrated Polyposis Syndrome. Clin Gastroenterol Hepatol. 2020 Apr;18(4):777–9.
33. van Leerdam M, Roos VH, van Hooft JE, Dekker E, Rover R, Kaminski MF et al. Endoscopic management of polyposis syndromes: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy 2019; 51: 877-95.
34. Bucksch K, Zachariae S, Ahadova A, Aretz S, Büttner R, Görgens H, et al. Adenoma and colorectal cancer risks in Lynch syndrome, Lynch-like syndrome and familial colorectal cancer type X. Int J Cancer. 2022 Jan;150(1):56–66.
35. Stjepanovic N, Moreira L, Carneiro F, Balaguer F, Cervantes A, Balmaña J, et al. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol. 2019 Oct;30(10):1558–71.
36. The International Mismatch Repair Consortium. Variation in the Risk of Colorectal Cancer for Lynch Syndrome. Lancet Oncol. 2021;2045(21):1–9.
37. Weiss JM, Gupta S, Burke CA, Axell L, Chen LM, Chung DC, et al. NCCN Guidelines® Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 1.2021. J Natl Compr Canc Netw. 2021 Oct;19(10):1122–32.
38. Gupta S, Provenzale D, Llor X, Halverson AL, Grady W, Chung DC, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal. Journal of the National Comprehensive Cancer Network. 2019 Sep;17(9):1032–41.
39. Møller P. The Prospective Lynch Syndrome Database reports enable evidence-based personal precision health care. Hered Cancer Clin Pract. 2020;18(1):1–7.
40. Idos G, Valle L. Lynch Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews® [Internet]. Seattle (WA); 2021.
41. Crosbie EJ, Ryan NAJ, Arends MJ, Bosse T, Burn J, Cornes JM, et al. The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genetics in Medicine [Internet]. 2019;21(10):2390–400. Available from: http://dx.doi.org/10.1038/s41436-019-0489-y
42. Hampel H, Pearlman R, Beightol M, Zhao W, Jones D, Frankel WL, et al. Assessment of Tumor Sequencing as a Replacement for Lynch Syndrome Screening and Current Molecular Tests for Patients With Colorectal Cancer. JAMA Oncol. 2018 Jun;4(6):806–13.
43. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (1979) [Internet]. 2017 Jul 28;357(6349):409 LP – 413. Available from: http://science.sciencemag.org/content/357/6349/409.abstract
44. Engel C, Vasen HF, Seppälä T, Aretz S, Bigirwamungu-Bargeman M, de Boer SY, et al. No Difference in Colorectal Cancer Incidence or Stage at Detection by Colonoscopy Among 3 Countries With Different Lynch Syndrome Surveillance Policies. Gastroenterology. 2018 Nov;155(5):1400-1409.e2.
45. Ahadova A, Seppälä TT, Engel C, Gallon R, Burn J, Holinski-Feder E, et al. The “unnatural” history of colorectal cancer in Lynch syndrome: Lessons from colonoscopy surveillance. Int J Cancer. 2021 Feb;148(4):800–11.
46. Biesalski HK. Gesunde Ernährung und Lebensstil und ihre Bedeutung in der Prävention von Krebs. Der Onkologe [Internet]. 2022;28(1):23–31. Available from: https://doi.org/10.1007/s00761-021-01046-y
47. Møller P. The Prospective Lynch Syndrome Database reports enable evidence-based personal precision health care. Hered Cancer Clin Pract. 2020;18(1):1–7.
48. Kastrinos F, Samadder NJ, Burt RW. Use of Family History and Genetic Testing to Determine Risk of Colorectal Cancer. Gastroenterology. 2020 Jan;158(2):389–403.
49. The International Mismatch Repair Consortium. Variation in the risk of colorectal cancer in families with Lynch syndrome: a retrospective cohort study. Lancet Oncol. 2021 Jul;22(7):1014–22.
50. Picó MD, Sánchez-Heras AB, Castillejo A, Giner-Calabuig M, Alustiza M, Sánchez A, et al. Risk of Cancer in Family Members of Patients with Lynch-like Syndrome. Cancers (Basel). 2020 Aug;12(8).
51. Rodríguez-Soler M, Pérez-Carbonell L, Guarinos C, Zapater P, Castillejo A, Barberá VM, et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology. 2013 May;144(5):924–6.
52. Ladabaum U, Dominitz JA, Kahi C, Schoen RE. Strategies for Colorectal Cancer Screening. Gastroenterology. 2020 Jan;158(2):418–32.
53. Alvarez MD, Quintana I, Terradas M, Mur P, Balaguer F, Valle L. The inherited and familial component of early-onset colorectal cancer [Internet]. Vol. 10, Cells. 2021. p. 710. Available from: https://www.mdpi.com/2073-4409/10/3/710
54. Terlouw D, Suerink M, Singh SS, Gille HJJP, Hes FJ, Langers AMJ, et al. Declining detection rates for APC and biallelic MUTYH variants in polyposis patients, implications for DNA testing policy. Eur J Hum Genet. 2020 Feb;28(2):222–30.
55. Yang J, Gurudu SR, Koptiuch C, Agrawal D, Buxbaum JL, Abbas Fehmi SM, et al. American Society for Gastrointestinal Endoscopy guideline on the role of endoscopy in familial adenomatous polyposis syndromes. Gastrointest Endosc. 2020 May;91(5):963-982.e2.
56. Aelvoet AS, Buttitta F, Ricciardiello L, Dekker E. Management of familial adenomatous polyposis and MUTYH-associated polyposis; new insights. Best Pract Res Clin Gastroenterol. 2022;58–59:101793.
57. Alvarez MD, Quintana I, Terradas M, Mur P, Balaguer F, Valle L. The inherited and familial component of early-onset colorectal cancer. Vol. 10, Cells. MDPI; 2021.
58. Macaron C, Mankaney GN, Haider M, Mouchli M, Hurley K, Burke CA. Chemoprevention Considerations in Patients with Hereditary Colorectal Cancer Syndromes. Gastrointest Endosc Clin N Am. 2022 Jan;32(1):131–46.
59. Burn J, Sheth H, Elliott F, Reed L, Macrae F, Mecklin JP, et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome),
PRAXIS
- Vol. 113
- Ausgabe 9
- Oktober 2024